Опубликовано в журнале:
«CONSILIUM MEDICUM»; ТОМ 8; № 8; стр. 80-87.Дисциркуляторная энцефалопатия: современные представления о механизмах развития и лечении
О.С.Левин
Кафедра неврологии Российской медицинской академии последипломного образования, МоскваПод дисциркуляторной энцефалопатией (ДЭП) принято понимать хроническую прогрессирующую форму цереброваскулярной патологии, характеризующегося развитием многоочагового или диффузного ишемического поражения головного мозга и проявляющуюся комплексом неврологических и нейропсихологических нарушений [1—3]. В отличие от ишемического инсульта, являющегося формой острой цереброваскулярной патологии, при которой обычно происходит фокальное поражение мозга, ДЭП характеризуется более постепенным развитием (часто с длительным периодом клинически «скрытого» течения), и мультифокальностью (диффузность) поражения мозга. Свойственная ДЭП тенденция к прогрессированию обычно связана с накоплением (кумуляция) полиморфных ишемических и вторичных дегенеративных изменений в мозге.
Эпидемиология
Широкая популярность концепции ДЭП среди практических неврологов в нашей стране и отсутствие четких критериев диагностики привели к явной гипердиагностике ДЭП, особенно у пожилых пациентов. Следует признать, что истинная распространенность хронической прогредиентной цереброваскулярной патологии остается неизвестной. Поскольку основным проявлением ДЭП является когнитивная дисфункция, ориентировочную оценку распространенности ДЭП можно сделать на основании проведенных в западных странах исследований распространенности сосудистых когнитивных расстройств. По данным различных авторов, когнитивные нарушения сосудистого генеза выявляются у 5-22% пожилых лиц [4, 5]. При аутопсии те или иные сосудистые изменения, чаще всего микроваскулярной природы, обнаруживают примерно у трети пожилых лиц. Таким образом, кумулятивная распространенность хронической цереброваскулярной патологии может составлять около трети пожилых лиц. Хотя сосудистая деменция, возникающая как в рамках ДЭП, так и после инсультов, уступает по распространенности болезни Альцгеймера, преддементные когнитивные нарушения сосудистого генеза легкой и умеренной выраженности, по-видимому, встречаются чаще, чем амнестический вариант умеренного когнитивного расстройства, рассматриваемый как продромальная фаза болезни Альцгеймера. Таким образом, если рассматривать весь спектр когнитивных нарушений (а не только деменцию), то цереброваскулярные заболевания, прежде всего ДЭП, могут быть их наиболее частой причиной по крайней мере у пожилых [6-8].Этиология и патогенез
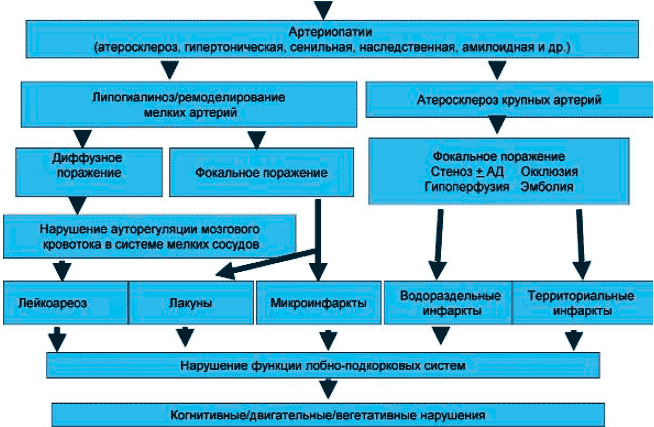
Клинические особенности ДЭП, по-видимому, можно объяснить тем, что в отличие от острых нарушений мозгового кровообращения большинство случаев ДЭП связано не с патологией крупных экстракраниальных артерий и их основных интракраниальных ветвей, а с поражением мелких мозговых артерий (церебральная микроангиопатия) [9, 10]. Основным этиологическим фактором церебральной микроангиопатии является артериальная гипертензия, вызывающая артериосклероз (липогиалиноз) мелких пенетрирующих артерий и артериол (гипертоническая артериопатия). У больных, не страдающих артериальной гипертензией, поражение мелких артерий может быть связано с сенильным артериосклерозом, амилоидной ангиопатией, значительно реже - с воспалительными или наследственными ангиопатиями (например, церебральная аутосомно-доминантная артериопатия с субкортикальными инфарктами и лейкоэнцефалопатией - ЦАДАСИЛ) [11]. Таким образом, как и инсульт, ДЭП представляет собой гетерогенное состояние, которое может иметь различную этиологию и по сути представляет собой клинический синдром.Распространенное поражение мелких артерий вызывает несколько основных типов изменений. Наиболее хорошо известны два из них:
1) диффузное двустороннее поражение белого вещества (лейкоэнцефалопатия),
2) множественные лакунарные инфаркты. Соответственно можно выделить лейкоэнцефалопатический (бинсвангеровский) вариант ДЭП, при котором выявляется диффузное поражение белого вещества (иногда в комбинации с лакунами), и лакунарный вариант ДЭП, характеризующийся наличием множественных лакунарных очагов. Если лакунарные очаги чаще обусловлены локальной окклюзией мелких артерий, то в генезе диффузного поражения белого вещества ведущая роль принадлежит повторяющимся эпизодам гипоперфузии, которые возникают в силу взаимодействия комплекса причин. Прежде всего из-за распространенной патологии микрососудов и системной артериальной гипотензии, которая может провоцироваться неадекватной гипотензивной терапией, ортостатической гипотензией вследствие вегетативной недостаточности, например при принятии вертикального положения или длительном стоянии, а также снижением сердечного выброса, например при пароксизмальных нарушениях сердечного ритма и т.д.Поражение мелких пенетрирующих сосудов, приводящее к диффузному поражению белого вещества, характеризуется не только их стенозом, но и, что не менее важно, их ареактивностью, в основе которой может лежать дисфункция эндотелия. Это приводит к нарушению ауторегуляции мозгового кровообращения, обеднению гемодинамического резерва и сужению «коридора» допустимых изменений перфузии. Из-за того, что в результате эндотелиальной дисфункции и последующего склероза, вызванными стойкой артериальной гипертензий или иными причинами, мелкие сосуды утрачивают способность расширяться, становится невозможным перераспределение перфузии в пользу активно работающих отделов мозга, а это в свою очередь приводит к их функциональной инактивации, а затем - и к необратимому повреждению. Преимущественное страдание белого вещества в перивентрикулярном и глубинных отделах при церебральной гипоперфузии объясняется особым характером их кровоснабжения, обеспечиваемого сосудами терминального типа, не имеющими коллатералей.
В результате хронической гипоперфузии или, что может быть более вероятным, повторных преходящих эпизодов гипоперфузии в глубинных слоях белого вещества полушарий развиваются так называемые неполные инфаркты, характеризующиеся демиелинизацией, гибелью олигодендроцитов, утратой аксонов, глиозом, но (в отличие от ишемического инсульта) не формированием очагов некроза. Кроме того, в зонах диффузного поражения белого вещества при патоморфологическом исследовании обнаруживают множественные мелкие инфаркты и кисты, расширение периваскулярных пространств с развитием etat crible, периваскулярный отек, валлеровская дегенерация, ангиоэктазии и другие изменения [1, 12]. Помимо гипоперфузии и ишемии в развитии этих изменений могут играть важную роль повторные эпизоды церебральных гипертензивных кризов, сопровождающиеся поражением сосудистого эндотелия, вазогенным отеком мозга, транссудацией плазменных белков и, возможно, токсических веществ, что ведет к периваскулярному энцефалолизису. Гибель структурных элементов белого вещества при недостаточном замещении образовавшихся дефектов астроцитами в тяжелых случаях приводит к формированию губчатой структуры белого вещества мозга (спонгиоз) [1].
В тех случаях, когда выявляются множественные лакунарные инфаркты в глубинных отделах мозга в отсутствие диффузного поражения белого вещества (лакунарный статус), процесс может быть связан с микроатероматозом начального отдела пенетрирующих артерий, идущих в глубь мозга, или закрытием атеросклеротической бляшкой просвета крупных сосудов в месте отхождения от него пенетрирующих ветвей.
Наряду с поражением глубинных отделов мозга у больных с церебральной микроангиопатией могут выявляться гранулярная атрофия корковых отделов и корковые микроинфаркты. Микроинфаркты - небольшие ишемические очаги до 5 мм в диаметре, выявляемые только при микроскопии. Они часто включают изменения, характерные для неполного инфаркта (снижение численности нейронов, аксонов, глиоз) и могут локализоваться как в коре, так и подкорковых структурах. Микроинфаркты могут быть связаны с артериолосклерозом, атеросклерозом крупных мозговых артерий, микроэмболией. Поражение крупных церебральных сосудов, основной причиной которого является атеросклероз, приводит к развитию более обширных (территориальных) корковых или подкорковых инфарктов и чаще бывает причиной инсультов, чем безинсультной ДЭП. В то же время при множественном атеросклеротическом стенозе крупных артерий возможно развитие прогредиентного ишемического поражения прежде всего в зонах смежного кровообращения (водораздельные зоны), находящихся на границе крупных сосудистых бассейнов. Морфологически в этих зонах могут выявляться ламинарный корковый некроз, микроинфаркты, неполные инфаркты и другие варианты селективной гибели нейронов (без формирования очагов некроза). В патогенезе поражения мозга при патологии крупных сосудов важное значение может иметь не только снижение перфузии, но и микроэмболизация [13]. Иногда ДЭП бывает результатом сочетанного поражения крупных и мелких мозговых артерий, соответственно при нейровизуализационном или патоморфологическом исследовании в этих случаях выявляется комбинация различных типов поражений. Схема развития ДЭП при артериальной гипертензии приведена на рисунке 1.
Патогенез дисциркуляторной энцефалопатии.Облигатным компонентом морфологической картины ДЭП является церебральная атрофия, которая может отражать наличие микроинфарктов, валлеровской дегенерации или непосредственно связана с гипоперфузией коры. У части больных церебральная атрофия отражает присоединение альцгеймеровских изменений в виде сенильных бляшек и нейрофибриллярных клубочков. Важными дополнительными факторами повреждения мозга при ДЭП, особенно при ее лейкоэнцефалопатическом варианте, являются сердечная недостаточность, приводящая к ограничению перфузии головного мозга, изменение реологии и свертываемости крови (например, вследствие распространенной эндотелиальной дисфункции, полицитемии, тромбоцитоза, гиперфибриногенемии, гиперлипидемии и т.д.); нарушение венозного оттока при стенозе или окклюзии глубинных мозговых вен или правожелудочковой недостаточности; апноэ во сне, вызывающие гипоксемию, нарушения сердечного ритма, колебания артериального давления; сахарный диабет; вторичные ликвородинамические нарушения.
Особенности клинических проявлений ДЭП определяются мультифокальным характером поражения мозга и преимущественным страданием его глубинных отделов, приводящим к разобщению корковых и подкорковых структур. В результате при ДЭП в наибольшей степени страдает функция лобных долей и их связей с подкорковыми и стволовыми отделами. Это предопределяет доминирующую роль когнитивных расстройств лобного типа и сложных нарушений двигательного контроля в клинической картине ДЭП.
Роль когнитивных нарушений в структуре клинических проявлений ДЭП
Хотя больные с ДЭП предпочитают акцентировать внимание на таких субъективных проявлениях, как головная боль, головокружение, шум в ушах, быстрая утомляемость, именно когнитивные нарушения следует признать ядром клинической картины ДЭП, которое в большинстве случаев определяет тяжесть состояния пациентов. Особенностью когнитивного дефицита у большинства больных ДЭП является преобладание нейродинамических и регуляторных когнитивных нарушений, связанных с дисфункцией соответственно I и III структурно-функциональных блоков по А.Р. Лурия, над нарушениями, связанными с дисфункцией II блока [2]. Это проявляется замедленностью психической деятельности, ослаблением внимания, снижением речевой активности, нарушением планирования, организации и контроля деятельности. Нарушение памяти, как правило, бывает умеренным и носит вторичный характер (об этом свидетельствует дефицит свободного воспроизведения при относительно сохранном узнавании и эффективности опосредующих приемов). Итогом прогрессирования нейропсихологических нарушений при ДЭП является развитие сосудистой деменции [14]. Однако ей предшествуют более легкие когнитивные нарушения, причем по мере прогрессирования когнитивного дефицита при ДЭП может происходить качественная трансформация его профиля. Нарастание и изменение профиля когнитивных нарушений позволяют оценить тяжесть ДЭП и отслеживать ее прогрессирование.Первой стадии ДЭП обычно соответствуют легкие когнитивные расстройства., преимущественно нейродинамические нарушения в виде замедленности, снижения работоспособности, истощаемости, колебаний внимания [15]. Тем не менее такие пациенты в целом хорошо справляются с тестами, не предусматривающими учета времени выполнения. Хотя подобные нарушения выходят за пределы возрастной нормы, они не ограничивают жизнедеятельности пациентов.
Второй стадии ДЭП чаще всего соответствуют умеренные когнитивные расстройства, которые наряду с нейродинамическими нарушениями включают и регуляторные нарушения (дизрегуляторный или подкорково-лобный когнитивный синдром). У таких пациентов нарушается выполнение даже тех нейропсихологических тестов, в которых не вводилось ограничение времени, но тем не менее сохраняется способность к компенсации когнитивного дефекта, что выражается в сохранном узнавании, эффективности опосредующих процедур, в частности «подсказок» в тестах на логическую память и абстрактное мышление. Подобный дефект полностью соответствует критериям умеренного когнитивного расстройства и, хотя не приводит к ограничению бытовой независимости пациента, может затруднять выполнение сложных (как правило, инструментальных) видов повседневной активности и способствовать снижению качества жизни больных [16].
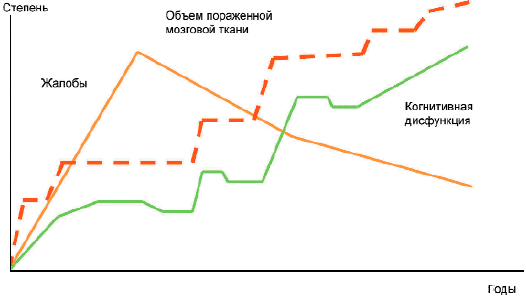
Третьей стадии ДЭП, как правило, соответствуют когнитивные нарушения, достигающие уровня деменции, т.е. нарушающие социальную адаптацию и хотя бы частично делающие пациента зависимым в быту от помощи окружающих. При деменции наряду с выраженными нейродинамическими и регуляторными нарушениями, которые остаются ядром когнитивного дефицита, отмечаются также операциональные нарушения, проявляющиеся в тестах на память, речь, праксис, зрительно-пространственные функции, мышление. В отличие от больных с умеренной выраженностью когнитивного дефицита на стадии 3 снижается эффективность узнавания и опосредующих процедур, предоставление пациенту подсказок или алгоритма действий в значительно меньшей степени улучшает выполнение тестов (рис.2).
Рис. 2. Динамика субъективных и объективных проявлений дисциркуляторной энцефалопатии.Приведенное соответствие между выраженностью когнитивных нарушений и стадиями ДЭП не является абсолютным.
Ведущая роль лобной дисфункции в структуре нейропсихологических нарушений проявляется также в нередком сочетании когнитивных и эмоционально-личностных нарушений. Последние на более ранних стадиях преимущественно представлены аффективными расстройствами (раздражительность, эмоциональная лабильность, тревожность, депрессия), на более позднем этапе к ним могут присоединяться выраженные личностные и поведенческие расстройства в виде апатико-абулических нарушений, расторможенности, эксплозивности, психотических расстройств и т.д.
Особенность двигательных нарушений при ДЭП
Хотя пирамидные знаки (оживление сухожильных рефлексов, анизорефлексия) встречаются у больных ДЭП довольно часто, парезы и спастичность наблюдаются сравнительно редко, если у больного отсутствуют эпизоды инсультов с острым развитием пирамидных нарушений. Постепенное развитие спастического пареза у больного с предполагаемой ДЭП требует исключения иного заболевания (спондилогенная шейная миелопатия, опухоли и т.д.). Тем не менее подострое (в течение нескольких недель) развитие гемипареза может быть связано с развитием стеноза или тромбоза внутренней сонной артерии (так называемый медленный инсульт). Мозжечковые и экстрапирамидные нарушения встречаются в струкутре клинических проявлений ДЭП нечасто. Гораздо чаще двигательные возможности пациента ограничиваются нарушениями ходьбы и равновесия, которые могут иметь комбинированный генез. Они бывают следствием поражения пирамидных, экстрапирамидных, мозжечковых систем, но нередко носят первичный характер и отражают нарушения функционирования сложных систем двигательного контроля, замыкающихся через лобную кору и включающих ее связи с подкорковыми и стволовыми структурами. Первичные нарушения ходьбы и равновесия, в зависимости от локализации и обширности поражения, могут быть представлены подкорковой (лобно-подкорковой) дисбазией, подкорковой или лобной астазией [17]. Именно сложные нарушения двигательного контроля наряду с псевдобульбарным синдромом и тазовыми нарушениями лучше всего коррелируют с выраженностью когнитивных нарушений.Клинико-нейровизуализационные корреляции в диагностике ДЭП
Термин «энцефалопатия» предполагает наличие не только субъективных жалоб, но и объективных признаков органического поражения мозга, которые могут быть обнаружены при неврологическом или нейропсихологическом исследовании. Вместе с тем обнаружение подобных признаков даже вкупе с сосудистыми факторами риска, клиническими или параклиническими признаками цереброваскулярной патологии является необходимым, но не достаточным признаком ДЭП. Важнейшим принципом диагностики ДЭП должна стать констатация причинно-следственной связи между имеющимися у больного клиническими проявлениями и цереброваскулярным заболеванием. Подобный принцип впервые был заложен в критерии клинической диагностики сосудистой деменции NINDS-AIREN. Представляется, что только следование этому принципу позволит избежать гипердиагностики ДЭП и отдифференцировать ДЭП от ряда нейродегенеративных заболеваний, широко представленных у лиц пожилого возраста (в первую очередь таких как болезнь Альцгеймера или болезнь Паркинсона).Доказательством причинно-следственной связи могут служить: особенности клинической картины (нейродинамический или дизрегуляторный характер когнитивного дефекта, сочетание когнитивных нарушений с аффективными нарушениями, а также неврологическими симптомами, свидетельствующими о страдании глубинных отделов мозга, в том числе дизартрией, экстрапирамидными знаками, нарушением ходьбы и др.), течение заболевания (постепенное начало с последующим неуклонным или ступенеобразным прогрессированием), соответствие клинической картины и данных дополнительных методов исследования в первую очередь компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ) головного мозга, отсутствием клинических или параклинических признаков другого заболевания, которое может объяснить клиническую картину [18, 19].
КТ или МРТ при ДЭП могут выявить: двустороннее более или менее симметричное диффузное поражение белого вещества (лейкоареоз) в перивентрикулярной зоне, зрительной лучистости, семиовальном центре; множественные лакунарные очаги (размером 3-15 мм) в базальных ганглиях, таламусе, мосте, мозжечке, внутренней капсуле, белом веществе лобных долей; более крупные корковые и подкорковые инфаркты, отражающие патологию крупных артерий. Церебральная атрофия, выявляемая при КТ или МРТ у больных ДЭП, обычно сопровождает лейкоареоз, лакунарые или территориальные инфаркты.
Как правило, расширение желудочковой системы при ДЭП более выражено, чем расширение корковых борозд, и может отражать не только убыль мозгового вещества в глубинных отделах мозга, но и, возможно, снижение резистентности перивентрикулярных тканей к ликвородинамическим воздействиям.
В ряде исследований выявлена связь между тяжестью и/или локализацией нейровизуализационных изменений и выраженностью когнитивных и двигательных нарушений [18]. Так, показано, что умеренное когнитивное расстройство возникает, когда распространенность лейкоареоза превышает как минимум 10% белого вещества полушарий, а деменция - если распространенность лейкоареоза превысит 1/4 объема белого вещества полушарий. При наличии лакун выраженность когнитивных нарушений зависит не столько от числа лакунарных очагов, сколько от их локализации (глубинные отделы лобных долей, головка хвостатого ядра и переднее бедро внутренней капсулы, таламус). Выраженность когнитивных нарушений увеличивается при двустороннем поражении указанных структур и сочетании лакунарных очагов с лейкоареозом. Более того, должно быть соответствие между нейровизуализационными изменениями и профилем когнитивных нарушений. Например, в отсутствие соответствующих корковых очагов на КТ и МРТ у пациентов не должны выявляться признаки очагового поражения корковых функций: афазии, апраксии и агнозии. Отмечена также связь между распространенностью лейкоареоза, особенно в передних отделах мозга, локализацией лакунарных очагов в чечевицеобразном ядре и выраженностью нарушений ходьбы и равновесия. Выраженность когнитивных и двигательных нарушений при ДЭП коррелирует и со степенью расширения боковых желудочков и особенно их передних рогов. С другой стороны, отсутствие сосудистых изменений на МРТ при клинической картине стадий 1-3 ДЭП и КТ при клинической картине стадий 2-3 ДЭП может заставлять усомниться в диагнозе. Данные КТ и МРТ имеют значение не только в диагностике ДЭП, но и могут помочь отслеживать динамику заболевания.
Сочетание ДЭП с нейродегенеративными заболеваниями
У пожилых лиц нередко наблюдается сочетание ДЭП с такими распространенными нейродегенеративными заболеваниями, как болезнь Альцгеймера или болезнь Паркинсона. Частое сочетание ДЭП и болезни Альцгеймера может объясняться наличием общих факторов риска (артериальная гипертензия, гиперхолестеринемия и т.д.) и, возможно, общих механизмов развития. Сосудистые и альцгеймеровские изменения могут оказывать аддитивное или синергическое действие, истощая церебральный резерв и способствуя клиническому проявлению друг друга. О наличии сопутствующей болезни Альцгеймера у больного ДЭП могут свидетельствовать нарастание когнитивного дефицита, связанного с дисфункцией височно-лимбических систем (плохое узнавание и опосредованное запоминание, низкий уровень семантически опосредованной речевой активности, раннее развитие зрительно-пространственных нарушений), развивающаяся атрофия гиппокампа по данным МРТ (см. таблицу 1). У больного с предполагаемой болезнью Альцгеймера о наличии ДЭП свидетельствуют не только выявляемые при КТ и МРТ очаговые или диффузные сосудистые изменения, но и раннее развитие нейродинамического и дизрегуляторного когнитивного дефицита, нарушения ходьбы и равновесия.Таблица 1
Сравнительная характеристика ДЭП, болезни Альцгеймера и их сочетания.
Признаки ДЭП Болезнь Альцгеймера ДЭП + болезнь Альцгеймера Сосудистые факторы риска (артериальная гипертензия, сахарный диабет, ожирение и т.д.) ++ ± + Признаки цереброваскулярного заболевания (транзиторные ишемические атаки или инсульт в анамнезе, стеноз сонных артерий и т.д.)
++ ±+ Течение Вариабельное (с периодами резкого ухудшения, частичного регресса или плато) Прогредиентное (возможны периоды плато) Прогредиентное (возможны периоды плато, резкого ухудшения, частичного регресса) Нейропсихологическое исследование Нарушение внимания и регуляторных функций
Нарушение опосредованного запоминания и узнавания в тестах на память
Снижение речевой активностиЗрительно-пространственные нарушения
++
±
В большей степени затрагивает ФА1 и ГА1Сравнительно поздно
±
++
В большей степени затрагивает СА1Рано
+
+/++
Вариабельное соотношение СА и ФАВариабельно
Аффективные нарушения + ± ± Двигательные нарушения (нарушения ходьбы, псевдобульбарный синдром, экстрапирамидные или пирамидные знаки) Проявляются рано Проявляются поздно Проявляются вариабельно Нейрогенные нарушения мочеиспускания Проявляются рано Проявляются поздно Проявляются вариабельно Данные МРТ атрофия гиппокампа множественные очаги/лейкоареоз2/
очаги в стратегических зонах3±
+++
±+
+1ФА - фонетические ассоциации (слова, начинающиеся на определенную букву, например, «л»), ГА - грамматические ассоциации (слова, относящиеся к определенной грамматической категории, например, глаголы), СА - семантические ассоциации (слова, относящиеся к определенной семантической категории, например, растения или животные).
2Распространенный перивентрикулярный/субкортикальный лейкоареоз, вовлекающий более 10% белого вещества полушарий.
3К стратегическим зонам относятся таламус, хвостатое ядро и другие базальные ганглии, лобная доля, угловая извилина, медиальные отделы височных долей.
± вариабельный признак, + признак, как правило, присутствует, ++ признак, как правило, значительно выражен.При присоединении ДЭП к болезни Паркинсона у больного может ускоренно нарастать когнитивный дефицит, дополнительно нарушается постуральная устойчивость, снижается эффективность противопаркинсонических средств.
Лечение ДЭП
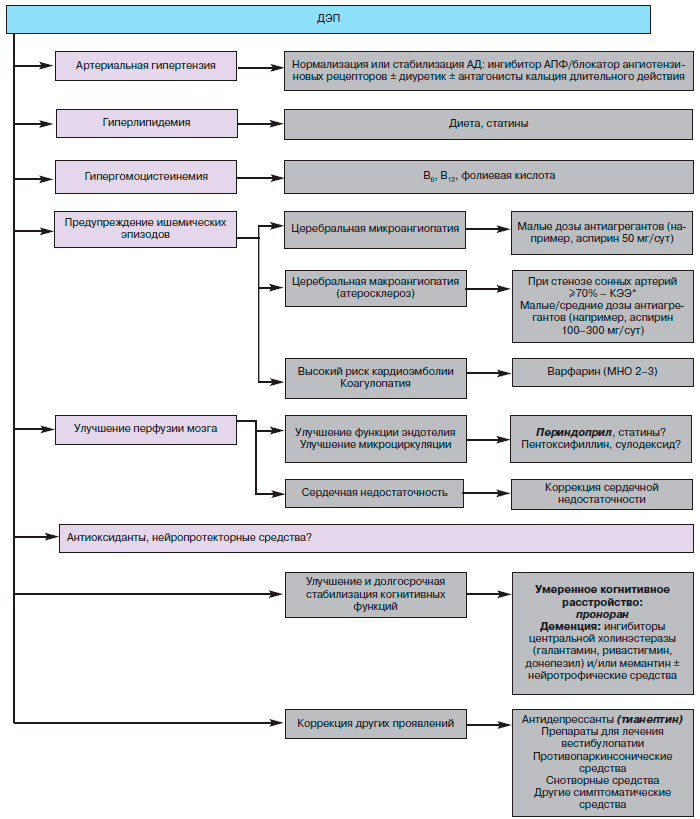
Лечение ДЭП должно иметь системный характер и включать меры по предупреждению дальнейшего повреждения мозговых сосудов и вещества мозга, улучшению и долгосрочной стабилизации когнитивных функций, коррекции других клинических проявлений заболевания (рис. 3). Наиболее эффективной мерой по предупреждению дальнейшего прогрессирования заболевания является воздействие на сосудистые факторы риска (артериальная гипертензия, сахарный диабет, гиперлипидемия, гипергомоцистеинемия, ожирение, курение). Особое значение имеет адекватная гипотензивная терапия. В ряде исследований показано, что гипотензивная терапия может замедлять развитие когнитивного дефицита. Так, в исследовании PROGRESS отмечено, что на фоне долгосрочного применения комбинации периндоприла («Престариум») и индапамида («Арифон») у больных, ранее перенесших инсульт, выраженность когнитивного дефицицита снижается на 16%, правда, главным образом за счет снижения риска повторных инсультов.
Алгоритм лечения дисциркуляторной энцефалопатии (КЭЭ – каротидная эндартерэктомия).В то же время коррекция артериальной гипертензии при ДЭП сложна ввиду того, что больным, начиная с определенного этапа, угрожает не только гипертензия, но и гипотензия, связанная в том числе и с приемом гипотензивных средств. Нестабильность артериального давления (АД) в дневное время, как и отсутствие ночного снижения АД, также неблагоприятно сказывающееся на состоянии мозгового кровообращения, могут быть также следствием самого заболевания, вызывающего дисфункцию центральных вегетативных структур. В начальный период артериальной гипертензии основная задача состоит в нормализации АД, предупреждающего повреждение сосудов. А с развитием когнитивных нарушений, особенно на фоне двустороннего стеноза магистральных артерий головы или выраженного повреждения системы мелких мозговых сосудов (на что может указывать обширный лейкоареоз или множественные лакунарные очаги), более важной становится задача стабилизации АД, возможно на несколько повышенном уровне, обеспечивающем оптимальную перфузию мозга в условиях нарушенной ауторегуляции мозгового кровообращения. У этой категории пациентов, по-видимому, следует стремиться к стабилизации АД на верхней границе нормы (систолическое АД должно поддерживаться на уровне 135-150 мм рт. ст.). Предпочтительнее использовать гипотензивные средства, в меньшей степени влияющие на мозговое кровообращение (оптимально назначение периндоприла, так как он не оказывает влияния на мозговой кровоток). Следует избегать приема антагонистов кальция короткого действия (например, нифедипин), провоцирующих резкие колебания АД. Но при необходимости можно использовать препараты антагонистов кальция пролонгированного действия.
Коррекция гиперлипидемии позволяет замедлить развитие атеросклеротического стеноза крупных мозговых артерий, снижает вязкость крови (что особенно важно при диффузном поражении мелких мозговых артерий), а также предупреждает прогрессирование ишемической болезни сердца. Статины, помимо снижения уровня холестерина, могут приводить к улучшению функции эндотелия, оказывать антитромбогенный и антиоксидантный эффекты, замедлять накопление в мозге β-амилоида.
У пациентов, перенесших инсульт или ТИА, а также имеющих выраженный атеросклеротический стеноз магистральных артерий головы и/или сосудистые очаги при КТ или МРТ, целесообразен длительный прием антиагрегантов. Препаратом первого выбора является аспирин, который назначают в дозе 50-300 мг 1 раз в день. При наличии противопоказаний к применению аспирина или его непереносимости возможна его замена на клопидогрель или добавление к аспирину дипиридамола. При коагулопатиях, постоянной форме мерцательной аритмии, других состояниях, связанных с высоким риском кардиогенной эмболии, антифосфолипидном синдроме показан прием антикоагулянтов. Антикоагулянты противопоказаны у лиц с лейкоэнцефалопатией в виду высокого риска внутримозговых геморрагий. У пациентов с атеросклеротическим стенозом сонных артерий при наличии легкого (но не тяжелого) когнитивного дефицита следует рассмотреть вопрос об оперативном лечении (каротидная эндартерэктомия, стентирование сонных артерий). При высоком уровне гомоцистеина показано назначение фолиевой кислоты, витаминов В6 и В12. Необходима адекватная коррекция сопутствующей соматической патологии, в частности сердечной и дыхательной недостаточности, гипотиреоза и т.д.
Улучшение кровообращения в системе мелких мозговых сосудов может быть обеспечено также с помощью препаратов, восстанавливающих функцию эндотелия (ингибитор АПФ с высокой тканевой специфичностью - периндоприл, статины), средств, улучшающих микроциркуляцию (например, пентоксифиллин), а также мерами, направленными на уменьшение вязкости крови (прекращение курения, коррекция гиперлипидемии или гиперфибриногенемии). До сих пор не удалось продемонстрировать в контролируемых испытаниях средств, предположительно поддерживающих метаболизм нейронов и оказывающих нейропротекторное действие. Несмотря на широкую популярность так называемых вазоактивных средств, их роль в лечении ДЭП окончательно не определена. Их способность в долгосрочном плане улучшать перфузию мозга не доказана. Более того, учитывая раннее снижение реактивности мелких сосудов в пораженных зонах мозга, на фоне применения вазоактивных средств возможен эффект обкрадывания в пользу интактных участков мозга с сохранными системами регуляции кровотока.
До сих пор не удалось подтвердить, что прием антиоксидантов (например, а-токоферола или др.) способен сдерживать прогрессирование когнитивного дефекта у больных с прогредиентным цереброваскулярным поражением.
Учитывая ключевую роль когнитивных нарушений в клинической структуре ДЭП, их коррекция имеет особенно важное значение. Для улучшения когнитивных функций применяется широкий спектр ноотропных препаратов, но в отношении большинства из них отсутствуют данные плацебо-контролируемых исследований, которые бы убедительно подтверждали их эффективность. Между тем, как показывают контролируемые исследования, клинически значимый эффект плацебо может отмечаться у 30-50% больных с когнитивными нарушениями, даже у пациентов с тяжелой деменцией.
На сегодняшний день у больных с уже развившейся сосудистой деменцией в контролируемых исследованиях показана эффективность ингибиторов холинэстеразы и модулятора глутаматных рецепторов. В среднем их эффективность следует расценить как умеренную. У больных с более ранней стадией ДЭП (при легких и умеренных когнитивных нарушениях) обнадеживающие данные получены при применении пирибедила.
Эффективность пирибедила (проноран) у больных с умеренным когнитивным расстройством, в том числе сосудистого генеза, показана в целом ряде исследований, в том числе проводимых с двойным слепым плацебо-контролем [20]. По данным D.Nagaraja и S.Jayashree (2001 г.), 3-месячный курс лечения пирибедилом в дозе 50 мг/сут вызвал улучшение когнитивных функций почти у 2/3 больных с умеренным когнитивным расстройством [21]. В контролируемом сравнительном исследовании пирибедил в дозе 50 мг/сут оказывал достоверно более выраженное положительное влияние на внимание, скорость реакции, память, речевую активность, аффективное состояние, чем винкамин в дозе 60 мг/сут [22]. G.Bartoli, E.Wichrowska (1976 г.) показали, что пирибедил в сравнении с плацебо приводит к достоверному уменьшению выраженности нарушений внимания, памяти и аффективных расстройств у пациентов с цереброваскулярной недостаточностью [23].
По данным В.В.Захарова и А.Б.Локшиной (2004 г.), у больных ДЭП с умеренным когнитивным расстройством пирибедил способствовал уменьшению выраженности когнитивных нарушений, связанных с лобной дисфункцией, параллельно уменьшая такие субъективные проявления, как головная боль, головокружение, снижение памяти, нарушения сна, утомляемость [24]. Результаты проведенного нами исследования эффективности пирибедила у 37 пациентов пожилого возраста с умеренным когнитивным расстройством показали, что препарат способствует улучшению нейродинамических функций, логической и зрительной памяти, речевой активности [25].
Особенностью нашего исследования была оценка эффективности пирибедила (пронорана) у пациентов с различным нейропсихологическим профилем умеренных когнитивных расстройств. Наибольшая эффективность отмечена при дизрегуляторном типе умеренных когнитивных расстройств, наименьшая - при амнестическом типе, при комбинированном типе умеренных когнитивных расстройств выявлены промежуточные результаты. Улучшение нейродинамических функций выявлено во всех трех группах, в то же время достоверное улучшение оценки по краткой шкале психического статуса, улучшение мнестических функций и речевой активности наблюдали лишь при дизрегуляторном типе. О роли хронической сосудистой мозговой недостаточности в развитии дизрегуляторного типа умеренных когнитивных расстройств свидетельствовало более высокое число сосудистых факторов риска, чем при комбинированном и амнестическом типах. Параллельно с улучшением когнитивного статуса отмечено снижение показателя когнитивных и общих жалоб, прежде всего за счет уменьшения жалоб на шум в ушах, головную боль, головокружение, общую слабость. Оценка по шкале депрессии Бека снизилась на 20%. По шкале EQ-5D к концу исследования отмечен достоверный положительный эффект по субшкалам, оценивающим повседневную активность, тревогу и депрессию.
Таким образом, пирибедил уменьшает выраженность умеренного когнитивного расстройства и сопровождающих их аффективных расстройств, связанных с начальными стадиями ДЭП. Механизм действия пирибедила связан с активацией D2/D3-дофаминовых рецепторов в лимбической системе, лобной коре, участках стриатума, регулирующих когнитивные процессы. В ряде исследований отмечено, что в основе когнитивной дифункции при цереброваскулярной патологии может лежать дисфункция дофаминергических систем [26]. Так, при сосудистой деменции снижается захват 18F-флуородопы и численность зон пресинаптического захвата дофамина в хвостатом ядре притом, что численность D2-рецепторов существенно не меняется [27, 28]. Со снижением активности дофаминергических систем мозга особенно тесно связаны снижение активности и концентрации внимания и другие проявления лобной дисфункции, сопровождающиеся вторичным расстройством памяти и аффективными нарушениями. По мнению G.Alexopoulos (2001 г.), аффективные нарушения, сцепленные с умеренным когнитивным дефектом лобного типа, относительно резистентны к традиционным антидепрессантам и могут лучше купироваться именно агонистами D3-дофаминовых рецепторов, к которым относится пирибедил [29]. Дополнительный механизм действия пирибедила может быть связан с блокадой α2-адренорецепторов, способствующей усилению норадренергической передачи в лимбической системе и лобной коры. Более того, как показывают экспериментальные данные, пирибедил, блокируя α2-адренорецепторы, приводит к усилению высвобождения ацетилхолина в лобной коре и дорсальной гиппокампе, что также может вносить вклад в усиление когнитивных функций [30]. Необходимы дополнительные исследования для оценки долгосрочного применения указанных средств на ранних стадиях ДЭП.
Важной задачей предупреждения деменции на этой стадии может быть формирование «когнитивного резерва». Решение этой задачи достигается не только применением средств, усиливающих когнитивные функции, но также адекватной умственной и физической нагрузкой, активной социальной деятельностью, методиками направленной нейропсихологической реабилитации.
При часто встречающихся тревожных невротических и неврозоподобных проявлениях необходима рациональная психотерапия в сочетании с антидепрессантами и прерывистыми курсами седативных препаратов и бензодиазепинов в малых дозах. При выраженной депрессивной симптоматике показаны антидепрессанты, предпочтительно не обладающие холинолитическим действием, например тианептин (коаксил). При лобной дисбазии с выраженным нарушением начала ходьбы и застываниями эффективна лечебная гимнастика, иногда некоторую пользу приносят также препараты амантадина, леводопы. При насильственном смехе и плаче применяют антидепрессанты. При вестибулярной дисфункции показана лечебная гимнастика, тренирующая вестибулярный аппарат и способность поддерживать равновесие, в сочетании с медикаментозными средствами, например бетагистином или никотиновой кислотой. При нарушении сна препаратами выбора являются агонисты бензодиазпиновых рецепторов, в резистентных случаях - малые дозы тразодона.
Заключение
ДЭП - одна из основных причин развития когнитивной дисфункции у пожилых. Раннее распознавание этого заболевания, включающее адекватную оценку нейровизуализационных изменений, комплексная терапия, основанная на современном понимании механизмов ее развития, могут создавать условия для сдерживания прогрессирования патологического процесса и долгосрочного улучшения качества жизни больных.ЛИТЕРАТУРА
1„ Верещагин Н.В., Моргунов В.А., Гулевская Т.С. Патология головного мозга при атеросклерозе и артериальной гипертонии. М.: Медицина, 1997.
2„ Сосудистые заболевания нервной системы. Под ред. Е.В.Шмидта. М.: Медицина, 1975.
3. Яхно Н.Н., „Дамулин И.В., Захаров В.В. Дисциркуляторная энцефалопатия. М., 2000.
4„ Grigsby J, Kaye K, Shertterly SM et al. Prevalence of disorders of executive cognitive functioning among elderly„ Neuroepidemilogy 2002; 21:213-20.
5„ Rockwood K, Wentzel C, Hachinscki V et al. Prevalence and outcomes of vascular cognitive impairment Neurology 2000; 54: 447-51.
6. Bowler JV, Hachinski V. The concept of vascular cognitive impairment. In TErkinjuntti, S.Gauthier (eds)„ Vascular cognitive impairment. Martin Dunitz 2002; p. 9-26.
7. Gauthier S, Touchon J. Subclassification of mild cognitive impairment in research and in clinical practice. Alzheimer's Disease and Related Disorders Annual 2004; p. 61-70.
8. O'Brien JT, Erkinjuntti T, Reisberg B et al. Vascular cognitive impairment. Lancet Neurology 2003; 2: 89-98.
9„ Дамулин ИВ, Брызжахина В.Г., Яхно НН. Нарушения ходьбы и равновесия при дисциркуляторной энцефалопатии Клинико-нейропсихологическое и МРТ сопоставление. Неврол. журн. 2004; 4:13-8.
10. Schmidtke K, Hull M. Cerebral small vessel disease: how does it progress?J Neurol Sciences 2005; 229/230: 13-20.
11. Roman GC, Erkinjuntti T, Wallin A et al. Subcortical ischemic vascular dementia. Lancet Neurology 2002; 1:426-36.
12.Левин О.С., Дамулин И.В.Диффузные изменения белого вещества и проблема сосудистой деменции. Достижения в нейрогериатрии. Под ред. Н.Н.Яхно И.В.Дамулина. М., ММА, 1995;189-228.
13. Локшина АБ, Захаров ВВ., Яхно НН. Легкие и умеренные когнитивные расстройства у пациентов с дисциркуляторной энцефалопатией. IX Всероссийский съезд неврологов, Ярославль, 2006; 69.
14. Reisberg B, Ferris S, Oo T et al. Staging: relevance for trial design in vascular burden of the brain. In TErkinjuntti, S.Gauthier (eds). Vascular cognitive impairment. Martin Dunitz 2002; p. 557-70.
15. Oswald WD, Tritt K. Cognitive deterioration in old age and in the course of dementia. In KAJellinger et al (eds). New trend in the diagnosis and therapy of Alzheimer's disease. Springer-Verlag, 1994; p. 105-14.
16„ Левин О.С, Сагова М.М., Голубева ЛВ. Факторы, влияющие на факторы жизни больных дисциркуляторной энцефалопатией с умеренным когнитивным расстройством. Рос. мед. журн. 2006; 2:
17. Левин О.С. Нарушения ходьбы: механизмы, классификация, принципы диагностики и лечения. В кн.: Экстрапирамидные расстройства. Под ред. В.Н.Штока и др. М.- Медпресс-информ, 2002; 473-94.
18. Яхно НН, Левин О.С,Дамулин ИВ. Сопоставление клинических и МРТ-данных при дисциркуляторной энцефалопатии. Когнитивные нарушения. Неврол. журн. 2001; 3:10-8.
19. Galluzzi S, Sheu C-F, Zanetti O et al. Distinctive clinical features of mild cognitive impairment with subcortical cerebrovascular disease. Dement Geriatr Cogn Disord 2005; 19:196-203.
20. Lambroso J, The value of Trivastal in the longterm treatment of chronic cerebral insufficience. CR Ther 1983; p. 1-9.
21. Nagaraja D, Jayaashree S. Randomized study of the dopamine receptor agonist piribedil in the treatment of mild cognitive impairment. Am J Psychiatry 2001; 158: 1517-9.
22. Scholing WE. A double-blind study using psychometric tests trivastal versus a reference compound. Tempo Medical 1982; 114b.
23. Bartoli G, Wichrowska E. Controlled clinical trial of piribedil in the treatment of cerebrovascular insufficiency. La Clin Terapeutica 1976; 78: 141-51.
24. Захаров В.В., Локшина А.Б., Применение препарата пирибедил (пирибедил) при легких когнитивных расстройствах у пожилых больных с дисциркуляторной энцефалопатией. Неврол. журн. 2004; 2:30-5.
25. Левин О.С., Голубева Л.В. Гетерогенность умеренного когнитивного расстройства: диагностические и терапевтические аспекты. Консилиум. 2006; 12:106-10.
26. Court J.A, Perry EK, Kalaria RN. Neurotransmitter changes in vascular dementia.J.O'Brien et al (eds). Cerebrovascular disease, cognitive impairment and dementia. London. Martin Dunitz, 2004; 133-52.
27. Allard P, Englund E, Marcusson JO. Reduced number of caudate nucleus dopamine uptake sites in vascular dementia. Dementia 1999; 10: 77-80.
28. Allard P, Englund E, Marcusson J. Caudate nucleus dopamine D2 receptors in vascular dementia. Dementia and geriatric cognitive disorders. 2002; 14:22-5.
29. Alexopoulos G. The depression-executive dysfunction syndrome of late life. Am J Geriatr Psychiatry 2001;9: 22-9.
30. Gobert A, DiCara B, Cistarelly L, Millan MJ. Piribedil enhances frontocortical and hippocampal release of acetylcholine in freely moving rats by blockade of a2A-adrenoreceptors.
| Октябрь 2012 г. |