Опубликовано:
Журнал неврологии и психиатрии, 7, 2009Особенности мониторинга и интенсивной терапии критических состояний при острых церебральных повреждениях
Д.м.н., проф., г.н.с. В.Г. Амчеславский
Peculiarities of monitoring and intensive therapy of critical states in acute cerebral damage
V.G.AmcheslavskyНИИ неотложной детской хирургии и травматологии, Москва
Ключевые слова: критические состояния, острые церебральные поражения, гипоксия мозга, первичное повреждение мозга, вторичное повреждение мозга, нейропротективная терапия, нейропротекторы.Key words: critical states, cerebral damage,
В 1979 г. Г.А. Рябовым [15] было дано следующее определение критического состояния: «..состояние больного, при котором наблюдаются расстройства физиологических функций и нарушения деятельности отдельных систем, которые не могут спонтанно корригироваться путем саморегуляции и требуют частичной или полной коррекции или замещения». Но всегда ли состояние, при котором производится коррекция или замещение физиологических функций и деятельности систем организма является критическим? Можно ли считать, например, критическим состояние больного в посленаркозном (послеоперационном) периоде? Является ли критическим состояние пациента на искусственной вентиляции легких (ИВЛ) с нарушением спонтанного дыхания после перенесенного полиомиелита? Можно ли считать критическим состояние больного с гиперазотемией при хронической почечной недостаточности? Во всех перечисленных случаях речь идет о нарушениях физиологических функций и деятельности органов, систем органов и гомеостаза, которые не могут корригироваться путем саморегуляции, что требует проведения заместительного лечения.
Сказанное говорит о том, что грань между описанными случаями и классически развивающимся критическим состоянием является пространственно-временнoй. Ее можно определить только с учетом остроты происходящего события и взаимосвязи первичного и последующих звеньев развития патологического процесса. Терминологически эта грань описывается словами — «острое расстройство функции, гомеостаза и т.д.», которое требует коррекции и «временного замещения» средствами и методами интенсивной терапии и реанимации. Так, у пациента с нарушениями дыхания после перенесенного полиомиелита, находящегося в зависимости от ИВЛ, критическое состояние может развиться после дисконнекции с респиратором, обусловившей развитие острой гипоксии.
Большой вклад в развитие проблемы критических состояний был внесен исследованиями А.З. Маневича и соавт. [8, 9], которые касались очаговых и диффузных повреждений головного мозга. Он дал определение критического состояния для таких случаев: «Критическое состояние при поврежденном мозге — это особое состояние больного, развитие которого обусловлено первичным или вторичным поражением структур мозга, ответственных за регуляцию системных и гуморальных механизмов жизнеобеспечения, что требует неотложного применения средств и методов интенсивной терапии и реанимации для контроля и управления временно нарушенными системными функциями и гомеостазом до полного или частичного восстановления центральной регуляции ими». В определенной мере это определение предвосхитило современную концепцию первичного и вторичного повреждения мозга, которая была опубликована в международных руководствах по лечению пострадавших с тяжелой черепно-мозговой травмой [28, 31].
Некоторые из аспектов формирования критических состояний (дыхание, гемодинамика, нейрогуморальный ответ, лечебно-охранительный режим и др.) при первичных церебральных повреждениях были уточнены в последующих исследованиях, [1, 2, 4, 10, 12, 14, 16, 17]. Важные результаты были получены благодаря современным методам нейровизуализации — компьютерной и магнитнорезонасной томографии [7].
Развитие первичного, приводящего к развитию церебрального повреждения под действием системных (внечерепных) факторов критического состояния, было описано и изучено в рамках общей реаниматологии. В соответствующих работах [3, 5, 6, 11, 15] на первом месте по значимости среди них стоят шок и гипоксия.
Что касается вторичного повреждения мозга, то оно интенсивно изучалось при тяжелой черепно-мозговой ским состояниям в остром периоде такой травмы А.А. Потаповым и соавт. [13].
Обобщению представлений о первичном и вторичном повреждении мозга в рамках концепции критических состояний во многом способствовали исследования при церебральных повреждениях другой этиологии (инсульт, острое субарахноидальное кровоизлияние — САК и др.).
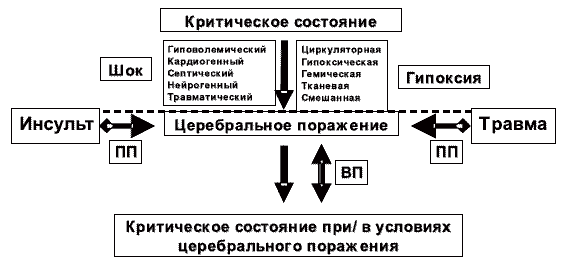
Таким образом, критическое состояние (вне зависимости от причин его возникновения) за счет действия повреждающих факторов может вызвать первичное поражение головного мозга. На первом месте среди таких повреждающих факторов критического состояния любой этиологии, стоят шок и гипоксия (рис. 1).
Рис. 1. Взаимозависимость критического состояния и церебрального повреждения.
ПП — первичное повреждение мозга (травма, инсульт, гипоксия, шок); ВП — вторичное повреждение мозга (ишемическое/гипоксическое под действием факторов вторичного повреждения мозга).Первичное поражение головного мозга вызывает нарушение его центрального регулирующего влияния на системные функции и гомеостаз. Это препятствует процессам стабилизации и восстановления системных функций и гомеостаза, преодолению критического состояния, несмотря на проводимое лечение. В следствие такого первичного повреждения головного мозга, происходит трансформация критического состояния в его особую форму. При этом факторы, определяемые нарушением системных функций и гомеостаза (нарушение системной гемодинамики, газообмена, метаболизма) действуют уже как факторы вторичного повреждения мозга, усугубляют тяжесть первичного церебрального поражения.
Первичное церебральное поражение может возникнуть и при исходно нормальном состоянии организма. Оно может быть травматическим (при ЧМТ), сосудистым (ишемический инсульт, кровоизлияние), гипоксическим (удушение, утопление). Оно может быть обусловлено непосредственным острым первичным повреждающим воздействием таких внутричерепных факторов, как опухоль, очаговая ишемия и/или кровоизлияние в мозг, нейрохирургическое вмешательство или остро возникающими и действующими извне повреждающими факторами (механическая энергия при ЧМТ, гипоксия и др.). Все это определяет последующее развитие критического состояния.
Особенности критического состояния в условиях первичного повреждения ЦНС определяют несколько составляющих: нарушение центральной регуляции системными и гуморальными механизмами жизнеобеспечения, обусловленное первичным церебральным поражением; риск усугубления первичного повреждения мозга под действием факторов вторичного повреждения, сопровождающих развитие и клиническое течение любого критического состояния; необходимость учитывать при диагностических и лечебных мероприятиях, проводимых в связи с критическим состоянием, что его развитие и/или поддержание обусловлено, в том числе, нарушением центральной регуляции жизненно важными функциями и гомеостазом.
Итак, первичное повреждение мозга — результат непосредственного воздействия на мозг повреждающих факторов: механической энергии при травме, внутричерепного кровоизлияния при геморрагическом инсульте; очаговой ишемии мозга при ишемическом инсульте; тотальной ишемии при циркуляторной недостаточности, гипоксии различного генеза, в том числе при шоке, и т.д. Естественно, возможно и сочетание таких факторов. Примером такого сочетания может быть ситуация, когда у водителя транспортного средства развился в ходе управления автомобилем инсульт с нарушением сознания, парезом/параличом, дезориентировкой и т.д., с потерей управления транспортным средством и последующим дорожнотранспортным происшествием, с получением тяжелой ЧМТ. Здесь первичное повреждение мозга определяют и инсульт и ЧМТ, то есть имеет место сочетание факторов первичного повреждения мозга (ишемия и воздействие механической энергии). Хотя они и связаны между собой взаимоотягощающими обстоятельствами возникновения, тем не менее, все они независимо повреждают мозг первично.
Первичное повреждение мозга является той данностью, с которой приходится иметь дело клиницисту при первичном осмотре больного. Повлиять на тяжесть первичного повреждения мозга врач не может. Более того, если пациент обследуется врачом в стационаре с позиций оценки тяжести первичного повреждения мозга, то, следовательно, тяжесть этого первичного повреждения уже совместима с жизнью больного на момент поступления в стационар. Выраженность первичного повреждения является той основой, которая определяет тяжесть состояния больного при госпитализации, клинический диагноз, стратегию и тактику неотложных мероприятий лечебнодиагностического характера. А вот от адекватности, своевременности, патофизиологической направленности, эффективности последних зависит то, в какой степени реализуется и будет выражено вторичное повреждение мозга.
Вторичное повреждение мозга — обусловлено действием вторичных факторов, в том числе тех, которые, сопровождают развитие критического состояния. Основная задача врача в этих случаях — выявление и уменьшение повреждающего действия таких факторов в условиях многопараметрового мониторинга и интенсивной терапии. От врача зависит, насколько может усилиться тяжесть первичного церебрального повреждения под действием факторов вторичного повреждения мозга, или насколько факторы вторичного повреждения мозга будут иметь самостоятельное танатогенное значение, определяя исход острого периода заболевания.
Условно выделяют внутричерепные и внечерепные факторы вторичного повреждения мозга. К внутричерепным факторам относят: присоединившиеся внутричерепные кровоизлияния и ишемию мозга, синдром внутричерепной гипертензии (ВЧГ), дислокационный синдром, церебральный вазоспазм и присоединившуюся внутричерепную инфекцию. Может возникнуть вопрос о первых двух из перечисленных внутричерепных факторов вторичного повреждения мозга, поскольку ранее они были отнесены к факторам первичного повреждения мозга. Но в данном контексте они являются присоединившимися, и потому вторичными. Например, у больного с геморрагическим инсультом очаг кровоизлияния оказывает компремирующее объемное воздействия на окружающие структуры мозга, вызывая компрессионную ишемию и отек тканей как проявление вторичного повреждения вещества мозга. Объемное воздействие очага кровоизлияния или ушиба мозга может привести к развитию ВЧГ и/или дислокационного синдрома, которые также относятся к внутричерепным факторам вторичного повреждения мозга. То же и в отношении развития внутричерепной инфекции, которая может быть как первичным повреждающим мозг фактором, так и присоединяться на каком-либо этапе после первичного повреждения мозга, например, при ЧМТ и усугублять тяжесть первичного повреждения, непосредственно участвуя в формировании критического состояния, влияя на прогноз и исход заболевания или травмы.
Внечерепные факторы вторичного повреждения мозга также хорошо известны клиницистам, к ним относят: нарушения газообмена, системной гемодинамики, температурной регуляции, углеводного и водно-электролитного (осмотического) гомеостаза, эндогенную интоксикацию и т.д. Свое повреждающее воздействие на мозг они оказывают на фоне уже состоявшегося первичного повреждения мозга.
В отличие от факторов первичного повреждения мозга, факторы вторичного его повреждения, как правило, сочетаются, взаимоотягощают свое повреждающее воздействие на мозг и могут возникать (присоединяться) неоднократно на протяжении всего острого периода заболевания.
В связи с этим необходим многопараметровый мониторинг состояния больного (пострадавшего) с момента первичной его оценки на месте происшествия, при транспортировке в стационар (догоспитальный период) и на всех этапах лечения в остром периоде заболевания (травмы). Такой мониторинг включает в себя комплекс методов непрерывной (продолженной) и дискретной (неоднократной) оценки наиболее важных параметров жизнедеятельности, функциональной и структурной целостности мозга и организма, гомеостаза и обменных процессов. Практически при всех критических состояниях в неврологии и нейрохирургии многопараметровой мониторинг определяется необходимостью выявления наиболее значимых факторов вторичного повреждения мозга.
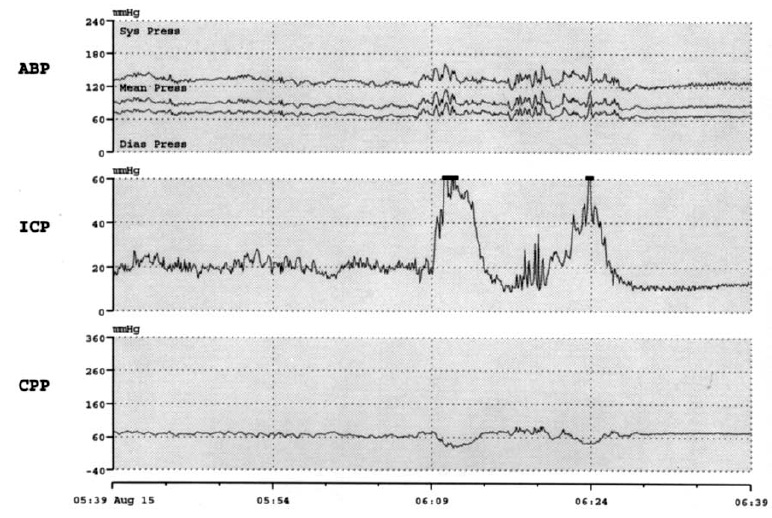
При ЧМТ одним из наиболее существенных, для исхода острого периода травмы является такой фактор вторичного повреждения мозга, как ВЧГ. Выраженность, длительность и резистентность ВЧГ к проводимому лечению в значительной степени определяют конечную эффективность всех усилий лечения и исход острого периода травмы. В связи с этим обязательным является включение в комплекс многопараметрового мониторинга методов контроля таких параметров, как внутричерепное давление (ВЧД) и церебральное перфузионное давление (ЦПД). Это обусловлено тем, что эпизоды повторного повышения ВЧД выше 20 мм рт.ст. длительностью более 30 мин могут развиваться на протяжении сравнительно коротких отрезков времени и вести к вторичному повреждению первично травмированного мозга (рис. 2), если они не выявлены и не предприняты меры их коррекции в рамках «пошаговой» стратегии борьбы с ВЧГ.
Рис. 2. Повторные патологические (>30 мин) эпизоды повышения ВЧД >20 мм рт.ст. соответствуют ухудшению исходов при тяжелой ЧМТ.
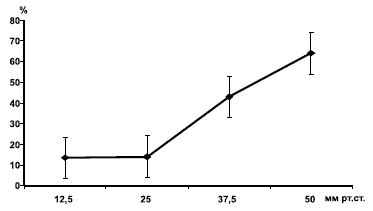
АВР — arterial blood pressure (артериальное давление), ICP — intracranial pressure (внутричерепное давление), CPP — cerebral perfusion pressure (церебральное перфузионное давление).Повторные эпизоды ВЧГ, по нашим данным и данным литературных источников, могут вести к значительному ухудшению результатов лечения пострадавших с тяжелой ЧМТ в остром периоде (рис. 3).
Рис. 3. Зависимость исходов острого периода тяжелой ЧМТ от эпизодов ВЧГ.
По оси ординат частота (%) неблагоприятных исходов (по шкале комы Глазго III—IV); по оси абсцисс — ВГД (мм рт.ст.),Показания к установке датчика для измерения ВЧД имеют четкий характер и основываются на сочетании результатов оценки клинических проявлений (оценка уровня сознания по шкале комы Глазго — ШКГ, наличие клинических признаков ВЧГ), данных нейровизуализационных исследований (в первую очередь КТ) и обстоятельств, обусловленных особенностями травмы (сочетанная, множественная) и ведения пострадавшего (массивная инфузионно-трансфузионная терапия).
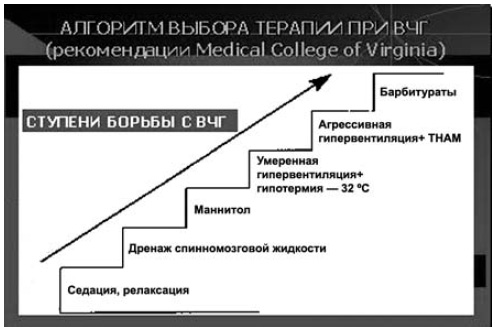
Для измерения ВЧД используют различные по стоимости системы, из которых наиболее доступной по цене и рекомендуемой по достигаемым клиническим эффектам (уменьшение ВЧГ) является измерение давления в боковых желудочках мозга при установке вентрикулярного дренажа. В этом случае используют систему жесткостенных (высокого давления) катетеров, соединенных с одной стороны с вентрикулярным дренажем, с другой с тензометрическим датчиком (используемым для измерения любого инвазивного давления — АД, центрального венозного давления — ЦВД соединенным, в свою очередь, через интерфейс монитора с блоком измерения инвазивного давления прикроватного монитора. Такой монитор, помимо блоков измерения частоты сердечных сокращений (ЧСС), насыщения кислородом капиллярной крови (SpO2), частоты дыхания (ЧД), температуры тела, должен иметь минимум 2 слота (гнезда) для блоков измерения инвазивного давления: один — для измерения ВЧД, другой — для измерения инвазивного АД. Такой вариант прикроватного монитора жизненно важных функций необходим, поскольку при одновременном измерении этих двух параметров (давлений) мы автоматически получаем результирующий (расчетный) параметр — ЦПД. Многочисленными исследованиями обоснована необходимость контроля этого параметра, поскольку он косвенно отражает состояние перфузии мозга, в том числе и адекватность (достаточность) пошаговой стратегии борьбы с синдромом ВЧГ. Критическим порогом снижения ЦПД для детей и взрослых считается уровень в 50 мм рт.ст. длительностью более 1 ч. Целесообразно поддерживать для взрослых ЦПД на уровне 70 мм рт.ст., а для детей 60 мм рт.ст. Именно на этом строится так называемая «пошаговая» стратегия борьбы с ВЧГ (схема 4), при которой каждый последующий «шаг» (ступень), является все более «агрессивным» по отношению к больному, связанным с возможностью развития побочных осложнений, и обусловлен (обоснован) неэффективностью предшествующего «шага» проводимой терапии синдрома ВЧГ.
Схема 4. «Пошаговый» (ступенчатый) алгоритм терапии при синдроме ВЧГ.
В схеме не указана первая ступень — возвышенное положение головы пациента (с нейтральным ее расположением во избежание компрессии шейных вен).В современных международных рекомендациях по лечению больных с тяжелой ЧМТ в остром периоде рассматривается отрицательное влияние и других факторов вторичного повреждения мозга на результаты лечения с позиций доказательной медицины. К ним относят, помимо ВЧГ, эпизоды снижения АД, эпизоды гипоксии, гипергликемию и др. Объединяют такие факторы следующие особенности их действия: 1) то, что они ведут к вторичному повреждению мозга; 2) ишемическигипоксический механизм повреждения — так называемый механизм вторичных мозговых повреждений (secondary brain insult»).
Существование универсального механизма вторичного повреждения мозга объединяет проблему острой травмы мозга с проблемой церебрального инсульта. Изучение последней на протяжении истекшего десятилетия позволило в максимальной степени определить механизмы нейронально-тканевого первичного повреждения, развивающегося при острых нарушениях мозгового кровообращения (ОНМК). Помимо единства механизмов повреждения клетки, развертывающихся при ее ишемии, вне зависимости от того, первично ишемическое повреждение на фоне ОНМК, или оно развилось на фоне факторов вторичного повреждения мозга, важно представлять этот процесс в динамике его развития, подразумевая единую временную шкалу. Если при ОНМК точкой отсчета является сам факт сосудистой катастрофы, а время, прошедшее с момента начальных ее проявлений, позволяет судить о стадии процесса, то при травме мозга ОНМК, как проявление действия факторов вторичного повреждения мозга, могут происходить и происходят на всем протяжении острого периода. Это, в свою очередь, предполагает одновременное наличие различных стадий ишемического каскада спустя уже несколько часов после травмы.
Суть процессов, происходящих в зоне ишемии, достаточно изучена, что позволяет утверждать, что в течение нескольких часов (до 6 ч) зона инфаркта окружена ишемизированной, но живой тканью — зоной «ишемической полутени», или пенумбры, в которой нарушен энергетический метаболизм, но имеются лишь функциональные, а не структурные изменения. Мозговая ткань в области «ишемической полутени» может быть возвращена к нормальной жизнедеятельности восстановлением адекватной перфузии ткани мозга, а применение нейропротективных средств способствует этому и пролонгирует период времени, в течение которого это возможно без дополнительного нарастания ишемических повреждений.
Интервалы времени, в течение которых происходят процессы клеточно-тканевого повреждения в зоне пенумбры при ишемическом инсульте, могут быть представлены в следующей последовательности: 0—3 ч — нарастающий энергетический дефицит; 3—6 ч — глутаматная эксайтотоксичность; нарушение Са2+ гомеостаза; нарастающий лактат — ацидоз; 12—36 ч — оксидантный стресс, воспаление; 48—72 ч — преобладание процессов апоптоза; позже 72 ч — необратимые морфофункциональные изменения в веществе мозга.
Именно зона пенумбры является главной мишенью терапии инсульта в первые часы и дни. Но если для ишемии при инсульте, как это уже отмечалось выше, можно представить нулевую точку отсчета этого временнoго интервала, то при ЧМТ вторичные ишемии могут развиваться на всем протяжении острого периода травмы. Таким образом, при ЧМТ мы имеем множественные ишемические повреждения на разных этапах их развития. При этом следует подчеркнуть, что механизмы вторичного повреждения головного мозга при внутримозговом и субарахноидальном кровоизлияниях так же, как и при ЧМТ, патогенетически имеют много общего с повреждением мозга при его ишемии (очаговой или диффузной) и гипоксии. Что, по сути, позволяет говорить о едином подходе к проведению нейропротективной терапии — защите мозга от повреждающих воздействий при всех этих состояниях.
При этом следует воспринимать нейропротективную терапию не только как медикаментозное воздействие, но и как совокупность всех существующих компонентов лечения с применением методов защиты мозга от действия факторов вторичного его повреждения. В связи с этим целесообразно выделение нескольких составляющих (подходов) в проведении нейропротективной терапии — физиологический, медикаментозный и хирургический подходы.
Физиологический подход к нейропротективной терапии
Физиологический подход к церебропротективной терапии подразумевает использование методов управления температурой тела, гемодинамикой, газообменом и гомеостазом. Речь идет об управляемых гипотермии, нормотермии, артериальной гипотензии, артериальной нормотензии, артериальной гипертензии, гиперволемии, гемодилюции, нормонатриемии, нормоосмолярности, нормовентиляции (гипервентиляции), управляемой нормогликемии (гипергликемия, гипогликемия)1.
Известны положительные эффекты гипотермии, которые легли в основу применения данной методики при острой гипоксии головного мозга. Лишь при данном типе острого церебрального поражения было с позиций доказательной медицины обоснованно применение гипотермии с понижением температуры вещества мозга до 32—34°С, как эффективного церебропротективного метода [32]. Это достигалось за счет: снижения скорости церебрального метаболизма кислорода; супрессии эксайтотоксичности; сохранения АТФ (аденозинтрифосфата) и ионного баланса нейронов; супрессии каскадов образования свободнорадикальных соединений; уменьшения ацидоза спинномозговой жидкости и продукции лактата; стабилизации клеточных мембран; восстановления функций гематоэнцефалического барьера. При большинстве других типов церебральных поражений, включая ЧМТ, положительное влияние гипотермии на результаты лечения не доказано. Во многом это связано с большим числом побочных эффектов и осложнений, обусловленных данным методом. К ним относят: кардиологические осложнения (аритмии, ишемия миокарда, снижение сердечного выброса из-за увеличения общего периферического сопротивления); коагулопатии; гипокалиемию; супрессию иммунной системы и увеличение риска инфекционных осложнений; гипергликемию; смещение кривой диссоциации оксигемоглобина (HbO2) влево.
В то же время, известно, что гипертермия является фактором вторичного повреждения мозга в остром периоде церебральных повреждений в связи, прежде всего, с ростом энергетических потребностей (на 8% с каждым 1°С), увеличением объемного мозгового кровотока и со ответствующим ростом ВЧГ [19]. В связи с этим рекомендуется поддержание нормотермии (не выше 37°С) методами физического охлаждения на фоне нейровегетативной блокады и применения антипиретиков.
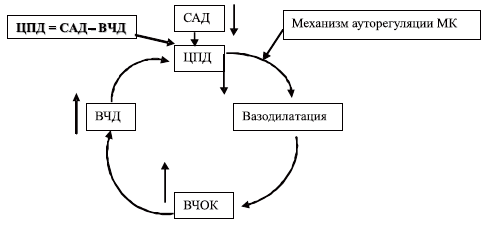
Артериальная гипотензия, а именно снижение систолического АД ниже 90 мм рт.ст., также является доказанным фактором вторичного повреждения головного мозга. Суть этого механизма повреждения мозга наглядно раскрывается концепцией вазодилатационного каскада М. Rosner и соавт. [40], изображенного на схеме 5.
Схема 5. Модель «комплексного вазодилаторного каскада».
САД — среднее артериальное давление, ВЧД — внутричерепное давление, ВЧОК — внутричерепной объем крови, ЦПД — церебральное перфузионное давление, МК — мозговой кровоток.Представленная модель иллюстрирует, как снижение ЦПД может стимулировать церебральную ауторегуляторную вазодилатацию с последующим повышением внутричерепного объема крови (ВЧОК) и ВЧД. Последняя величина, являясь интегральным показателем состояния внутричерепных объемных соотношений, возрастает при увеличении ВЧОК согласно концепции Монро-Келли, как отражение возрастания внутричерепных объемов и исчерпания резервов компенсации внутричерепного содержимого за счет перераспределения этих объемов. Если среднее артериальное давление (САД) не изменяется, то ЦПД снизится дальше, и цикл будет продолжаться до достижения максимальной дилатации мозговых сосудов или до соответствующего изменения САД. Этот каскад может быть инициирован в любой точке. Например, гипоксемия может стимулировать начало вазодилатации с последующим запуском каскада. Лекарственные препараты, дегидратация, шокогенные факторы, влияющие на САД, могут инициировать каскад на системном уровне [40].
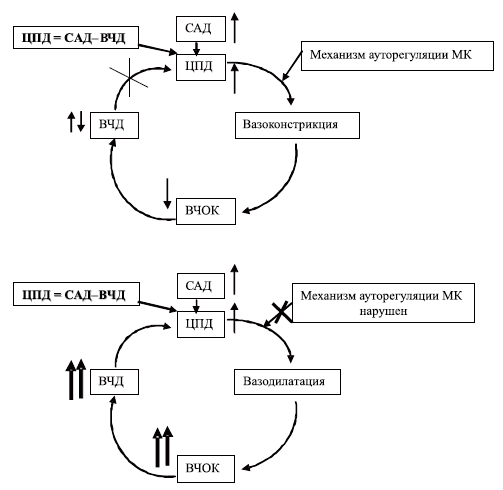
Согласно данной концепции для предупреждения дальнейшего снижения ЦПД необходимо повышать и поддерживать САД на определенном уровне, позволяющем обеспечить требуемую величину ЦПД. В случае, когда ЦПД повышается, развивается ауторегуляторная вазоконстрикция, снижаются ВЧОК и степень выраженности ВЧГ, таким образом, вазодилатационный каскад прерывается (схема 6 I). Рекомендуется поддерживать ЦПД у взрослых не менее 70 мм рт.ст., а у детей не менее 60 мм рт.ст., используя методы управления САД (гипердинамией — катехоламины, вазопрессоры, другие сердечнососудистые средства и гиперволемией — коллоиды, кристаллоиды). На этом принципе основана так называемая целевая по должному ЦПД терапия церебральной ишемии при ВЧГ у больных с тяжелой ЧМТ.
Схема 6. Изменение модели «комплексного вазодилаторного каскада» при повышении ЦПД (I) и нарушении механизма ауторегуляции МК (II).Легко представить, что для поддержания ЦПД на уровне 70 мм рт.ст. при ВЧД равном 20 мм рт.ст., необходимо достичь САД 90 мм рт.ст. То есть АД пациента должно быть не менее 130/70 мм рт.ст. Это исходит из формулы расчета ЦПД: ЦПД=САД–ВЧД, где 70 (ЦПД)=[130 (АД систолическое)+140 (2•АД диастолическое)]/3—20 (ВЧД). То же можем пересчитать с точки зрения рекомендуемой для детей величины ЦПД=60 мм рт.ст.: 60=(100+2•70)/3—20. То есть требуемые цифры АД ребенка должны быть 100/70 мм рт.ст.
Безусловно, при проведении мониторинга, включающего прямой (инвазивный) контроль АД и прямой инвазивный контроль ВЧД, величина ЦПД рассчитывается автоматически и выводится на дисплей прикроватного монитора жизненно важных функций. Только в этих условиях возможно проведение «пошаговой» стратегии борьбы с ВЧГ, которая, по сути, является терапией, направленной на поддержание ЦПД путем управляемой артериальной гипертензии.
Всегда ли подобная терапия адекватна задаче уменьшения выраженности ВЧГ? Оказывается, нет. Представим себе, что тяжесть ЧМТ такова, что обусловливает нарушение ауторегуляции мозгового кровотока (см. схему 6II). В этом случае, просто повышая АД, мы вызовем пропорциональное возрастание ВЧОК, так как отсутствует механизм ауторегуляторной вазоконстрикции. Такое возрастание ВЧОК приведет к быстрому росту ВЧД и вновь к замыканию круга патологических реакций: повышение АД — увеличение ВЧОК — рост ВЧД — отсутствие эффективного повышения ЦПД — вновь повышение АД. Мы просто не сумеем получить достаточное увеличение ЦПД в связи с постоянным ростом ВЧД, практически пропорциональным увеличению АД. У таких пациентов эффективной является терапия, направленная на снижение ВЧД — так называемая, целевая, по должному ВЧД (ICP — “target” therapy) терапия.
Альтернативой подобному варианту лечения является концепция управляемой артериальной гипотензии. Эта концепция, вышедшая в свет из стен клиники университета г. Лунд (Швеция), носит название Лундовской концепции и обосновывает допустимость и целесообразность снижения ЦПД у взрослых до 50 мм рт.ст., а у детей до 30—40 мм рт.ст. [27].
При проведении лечения согласно данной концепции важно учитывать необходимость: поддержания нормального коллоидно-осмотического давления (переливанием компонентов крови и белковосодержащих растворов); снижения внутрикапиллярного гидростатического давления, применяя β1-антагонисты (метопролол)+ α2агонисты (клонидин) + прекапиллярные вазоконстрикторы (дегидроэрготамин, малые дозы тиопентала натрия); уменьшения внутричерепного объема крови (артериальной — дегидроэрготамином, малыми дозировками тиопентала натрия и венозной — дегидроэрготамином, применением венотоников, улучшая венозный отток из полости черепа, снижая венозное давление).
Для обоснованного применения такой терапии необходимо точно установить, существует или нарушен механизм ауторегуляции мозгового кровообращения (МК). Косвенно об этом можно судить исходя из тяжести травматического повреждения мозга, поскольку нарушение ауторегуляции МК сопутствует наиболее тяжелым вариантам травматического повреждения. Например, нередко нарушение ауторегуляции МК отмечают у больных, перенесших дислокационный синдром с ущемлением мозгового ствола, у больных с диффузным аксональным повреждением мозга III—IV степени тяжести.
С точки зрения клинических проявлений следует отметить, что нарушению ауторегуляции МК соответствует выявляемое в ходе многопараметрового мониторинга возрастание ВЧД, пропорциональное повышению АД, при одновременном снижении ЦПД. Тем не менее наиболее объективным критерием такого нарушения является совокупность вышеизложенного в сочетании с определением резерва динамической миогенной ауторегуляции МК по коэффициенту овершута (КО)2.
Выбор той или иной тактики в рамках физиологического подхода к проведению нейропротективной терапии определяется врачом на основании комплексной оценки состояния больного, включающей данные многопараметрового мониторинга и инструментальных методов обследования. Важно помнить о том, что при отсутствии возможности дифференцированной оценки состояния пациента, в частности, церебральной гемодинамики, с позиции выбора одной из перечисленных тактик ведения пациента следует придерживаться международных рекомендаций, а именно, предупреждать эпизоды снижения систолического АД ниже 90 мм рт.ст. [28, 31] всеми доступными методами.
Нарушения водно-электролитного обмена могут быть фактором вторичного повреждения мозга и представлены в остром периоде церебральных повреждений различной этиологии двумя основными вариантами: формированием гиперосмолярно-гипернатриемического синдрома (ГГС) и гипоосмолярно-гипонатричемическими нарушениями.
ГГС (повышение натрия выше 150 ммоль/л и осмолярности выше 320 мосмоль/л) наиболее часто развивается, сопутствует и осложняет острый период тяжелой ЧМТ, геморрагического инсульта, постгипоксической энцефалопатии. Это, как правило, является отражением нарушения целостности и функции структур гипоталамогипофизарной оси (центральных звеньев) и дистальных канальцев, собирательных трубочек почек (эффекторных звеньев), являющихся в норме единой системой регуляции водно-солевого гомеостаза. Гиперосмолярность мозговой ткани может сформироваться и при длительном или неадекватном (непрерывном) введении осмотически активных препаратов (маннитол, мочевина), когда происходит их постепенное накопление в ткани мозга. Развившись, ГГС ведет к метаболической недостаточности клетки и, в конечном итоге, к развитию метаболического отека, связанного с избыточным поступлением и накоплением метаболитов в клетках с увеличением их объема. Все это вместе ведет к усилению энергетической недостаточности клетки, ее гипоксии, вторичному повреждению мозга. Сложность клинической ситуации заключается еще и в том, что быстрая коррекция гиперосмотических, гипернатриемических нарушений может приводить к другим осложнениям (осмотическому отеку и росту ВЧГ, дислокационному синдрому, метаболическим нарушениям). Чем выраженнее нарушения при ГГС, тем большее время необходимо для их постепенной коррекции. То же относится и к гипонатриемическим, гипоосмолярным нарушениям, которые чаще являются последствием ятрогенных воздействий (переливание гипотоничных растворов, быстрое снижение глюкозы при гиперосмолярном кетоацидозе) или синдрома несоответствующей секреции антидиуретического гормона (SIADH). Нарастающий их характер, а также быстрая коррекция гипонатриемии и гипоосмолярности могут приводить, помимо перечисленных выше (нарушений), к синдрому центрального миелинолиза моста и подкорковых структур.
Уменьшить выраженность вторичного повреждающего воздействия водно-электролитных нарушений на мозг возможно с учетом представления универсальных механизмов этого повреждения (энергетическая клеточная недостаточность, гипоксия клетки), используя средство многоцелевого воздействия — актовегин. Его возможность оптимизировать и энергообмен, и газообмен клетки будет рассмотрена далее, здесь лишь отметим, что его фоновое применение в дозировке до 20 мг/кг массы тела увеличивает резерв времени для проведения коррекции водно-электролитных нарушений без опасности нарастания вторичного повреждения мозга. Тем не менее рекомендуется в остром периоде церебральных повреждений поддерживать уровень нормонатриемии и нормоосмолярности для избежания вторичного повреждения мозга.
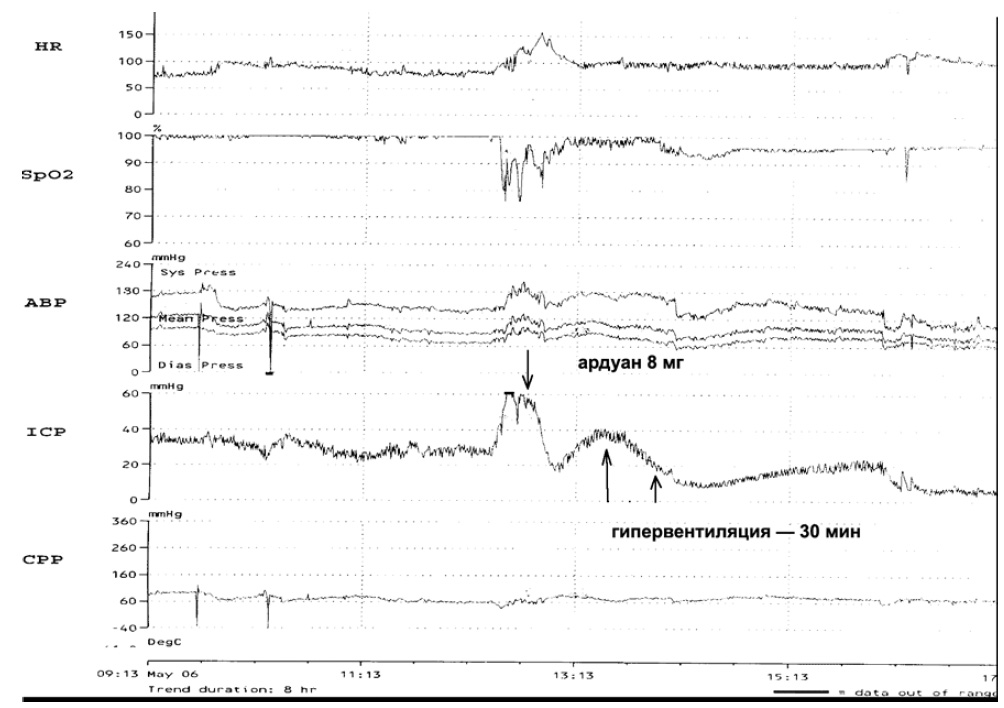
Нарушения газообмена, в том числе связанные с респираторными нарушениями, известный фактор вторичного повреждения мозга. На рис. 7 приведен пример того, как десинхронизация больного с респиратором (гипоксемия, рост внутригрудного давления, нарастание гиперкапнии) ведет к повышению ВЧД.
Рис. 7. Десинхронизация у больного с респиратором ведет к росту ВЧД. Применение миорелаксанта (ардуан) и последующая кратковременная гипервентиляция позволили нормализовать ВЧД.
HR — частота сердечных сокращений, SpO2 — насыщение кислородом капиллярной крови, АВР — артериальное давление, ICP — внутричерепное давление, CPP — церебральное перфузионное давление.Применение гипервентиляции и мышечных релаксантов позволяет нивелировать нарушения газообмена и респираторной механики со снижением ВЧД. Подобный вариант управления внутричерепными объемными соотношениями путем миорелаксации и гипервентиляции традиционно применяется при анестезиологореанимационном обеспечении больных с церебральными поражениями различной этиологии. Например, для облегчения доступа при нейрохирургических операциях в условиях ВЧГ. Релаксация больного является первым «шагом» при выполнении пошаговой стратегии борьбы с ВЧГ у пострадавших в остром периоде тяжелой ЧМТ.
Методика гипервентиляции обоснована суммой ее положительных влияний на внутричерепные объемные соотношения, доставку кислорода, элиминацию углекислоты, нормализацию кислотно-основного состояния с коррекцией лактацидоза [24, 25, 35].
В то же время гипервентиляция и гипокапния, связанная с ее проведением, вызывают ряд неблагоприятных эффектов. Наиболее значимым осложнением пролонгированной (более 1 ч) гипервентиляции является развитие церебральной ишемии в результате рефрактерного вазоспазма, а также усугубление существующих очагов ишемии за счет снижения коллатерального кровотока [33]. Помимо ишемии мозговой ткани, развивающийся ишемический, а далее вазогенный отек, ведут к нарастанию ВЧГ. Особенностью отека мозга при рефрактерном вазоспазме является невозможность купирования отека и нарастающей ВЧГ продолжением гипервентиляции, меньше и эффективность осмотических диуретиков, что существенно снижает возможности борьбы с нарастающим ВЧД. Поэтому рекомендуется поддерживать баланс нормокапнии управляемой респираторной терапией, при необходимости седатацией и релаксацией, адекватной нутритивной поддержкой.
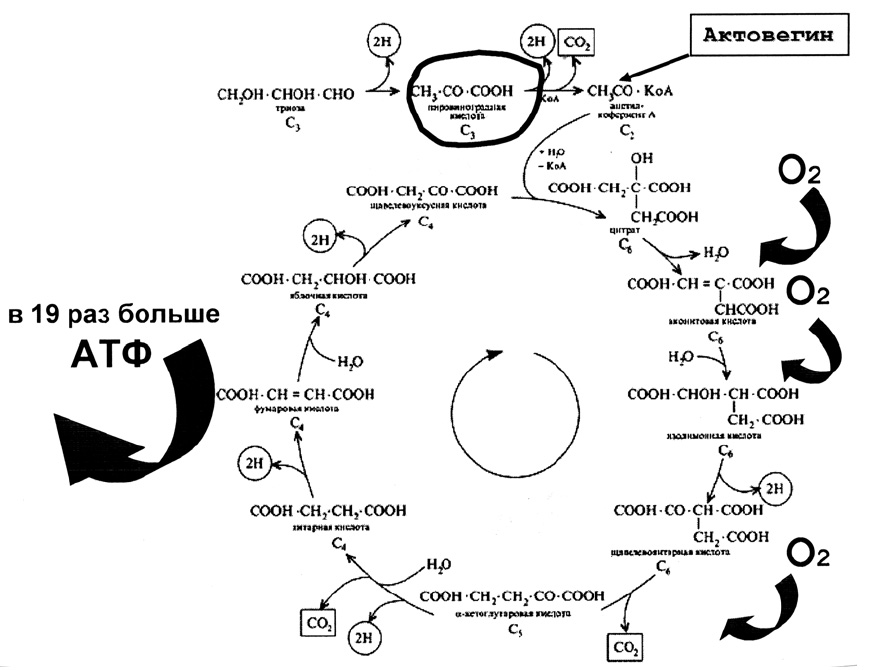
Оптимизации процессов клеточно-тканевого дыхания можно достигнуть и фоновым применением препарата актовегин, поскольку он физиологическим путем (увеличивая активность естественных ферментов окислительного фосфорилирования, в первую очередь, сукцинатдегидрогеназы и пируваткиназы) стабилизирует клеточное дыхание (рис. 8). Актовегин способствует переходу анаэробного пути окисления в аэробный, достигая увеличения выработки АТФ в 19 раз (!) и снижает, тем самым, образование лактата и степень выраженности лактацидоза.
Рис. 8. Механизм действия актовегина на ключевые звенья цикла Кребса (увеличение выработки АТФ).Нарушения углеводного обмена являются фактором вторичного повреждения мозга. Гипогликемия не только при острых церебральных повреждениях, но и без них, развиваясь в силу различных причин, может приводить к развитию гипоксически-ишемических нарушений в мозге, что нередко проявляется синкопами нарушения сознания, острой слабости, артериальной гипотонии. На клеточном уровне это является следствием разобщения процессов окислительного фосфорилирования, на системном — гипокапнии.
Тем не менее при острых церебральных повреждениях чаще отмечается развитие гипергликемических состояний. Это связано со многими факторами, сопутствующими острому периоду заболевания, будь то ЧМТ или церебральный инсульт. К этим факторам относят выброс контринсулярных гормонов, мобилизацию эндогенных запасов глюкозы, процессы неоглюкогенеза, формирование инсулинорезистентности.
Из перечисленных факторов именно формирование инсулинорезистентности представляет наибольшую проблему с точки зрения коррекции гипергликемии. В условиях формирования инсулинорезистентности, даже при достаточной секреции эндогенного инсулина, требуется введение дополнительных его количеств парентерально. Это, в свою очередь, приводит к образованию аутоантител к инсулину, что усугубляет процесс инсулинорезистентности и требует еще больших количеств экзогенного инсулина. Следует помнить, что повышенное содержание глюкозы в крови в условиях инсулинорезистентности сопровождается сниженным поступлением ее в клетку, что является основой энергетической недостаточности, характерной для этого периода заболевания. А это сниженное количество глюкозы в клетке расщепляется преимущественно по анаэробному пути в связи с недостаточным количеством поступающего кислорода, что усугубляет энергетическую недостаточность и лактацидоз, о чем было сказано выше.
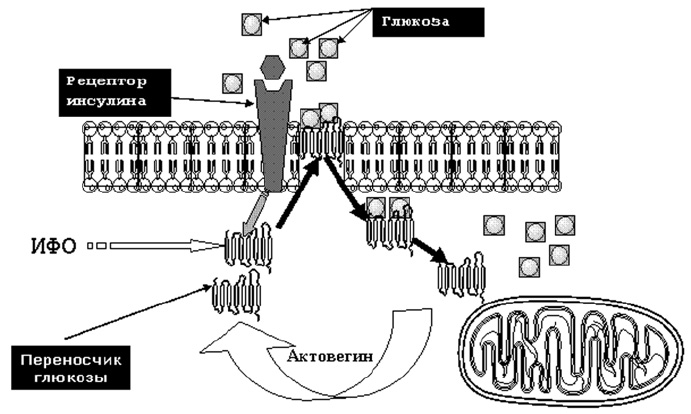
Также важно помнить и то, что в организме человека существует 3 типа инсулиннезависимых клеток — гепатоциты, эритроциты и нервные клетки. Если у эритроцитов инсулиновые рецепторы отсутствуют полностью, то у гепатоцитов и нервных клеток они хотя и имеются, но в обычных условиях функционально не активны. Поступление глюкозы в клетки этих типов происходит альтернативным путем с помощью так называемых переносчиков глюкозы. Такой путь поступления глюкозы в клетки называется инсулиннезависимым. Стимуляция поступления глюкозы в клетку при инсулиннезависимом пути происходит за счет активации инозитолфосфатолигосахаридов (ИФО). Именно путем воздействия на ИФО (GLUT — 1,5) реализуется фармакологический эффект актовегина, способствующего улучшению поступления глюкозы в нервную клетку физиологическим путем (рис. 9). На этом же механизме основано улучшение поступления глюкозы в клетку при таком варианте инсулинорезистентности, который имеет место при сахарном диабете и диабетической полиневропатии.
Рис. 9. Механизм инсулинонезависимого действия актовегина в ткани мозга.Таблица
Мишени Средства воздействия Нарушение обеспечения нейрона энергией Макроэрги, метаболики (актовегин, неотон, цераксон) Развитие отека мозга Осмотические диуретики Ацидоз ТНАМ (трометамин) Процессы перикисного окисления Индометацин, 21-аминостероиды, цераксон Избыток свободных радикалов Антиоксиданты (актовегин, токоферол, цераксон, глицин, мексидол) Эксайтотоксическое действие нейротрансмиттеров Антагонисты нейротрансмиттеров (глицин и др.) Инфлюкс Са2+ в нейрон Блокаторы Са2+ каналов (нимодипин) Демиелинизация, нарушение синаптической передачи Донаторы холина (цераксон), глиатилин и др. антихолинэстеразные средства
Снижение глютамат-индуцированного апоптоза (цераксон)Тем не менее, в остром периоде церебральных повреждений рекомендуется поддержание нормогликемии, что достигается адекватной нутритивной поддержкой, контролем уровня гликемии с коррекцией поступления глюкозы, прежде всего, в нервную клетку.
Поддержание уровня нормогликемии, в том числе и адекватного поступления глюкозы в клетку, в этих условиях существенно облегчается фоновым применением актовегина, что является примером того, как препарат целевой (дифференцированной) терапии может с полным основанием применяться как средство базисной терапии.
Медикаментозный подход к нейропротективной терапии
Медикаментозный подход к церебропротекции является наиболее интенсивно развивающимся направлением в клинической неврологии. В настоящее время выделяются многочисленные мишени — ключевые звенья и механизмы нарушений, развивающихся при гипоксии клетки, соответственно которым созданы и применяются, а также продолжают создаваться многочисленные медикаментозные средства.
Приведен далеко не полный перечень мишеней и средств медикаментозного воздействия на них. Обилие лекарственных препаратов на фармакологическом рынке определяет проблему дифференциального выбора, показаний и противопоказаний к их назначению. К сожалению, большинство препаратов не соответствуют требованиям доказательных исследований их эффективности. Поэтому практически речь идет о применении того минимума препаратов, эффективность которых показана при стандартных клинических ситуациях методами доказательной медицины.
Возвращаясь к терапии, называемой «пошаговой» стратегией борьбы с ВЧГ, мы обязательно приводим осмодиуретик маннитол, как «доказанное» средство борьбы с ВЧГ. Маннитол снижает ВЧД за счет реализации 2 механизмов действия: быстро развивающегося, но краткосрочного — увеличение объема циркулирующей плазмы (гиперволемический эффект мобилизации жидкости) и уменьшение вязкости крови согласно механизму ауторегуляции мозгового кровообращения, что приводит к сужению сосудов мозга, уменьшению ВЧОК и снижению ВЧД. Более длительное снижение ВЧД обусловлено осмотическим эффектом маннитола, уменьшающего внеклеточный объем за счет выхода жидкости из внеклеточного пространства в сосудистое русло по осмотическому градиенту. Помимо этого, маннитол уменьшает ригидность эритроцитов, что позволяет им проходить через сосуды малого калибра к зонам с краевой перфузией; является «ловушкой» свободных радикалов, снижает скорость образования цереброспинальной жидкости. Эти эффекты маннитола реализуются при правильном его применении. Маннитол следует вводить болюсами в течении 20—30 мин в дозировке — 1,0 г/кг массы тела взрослым и 0,5 г/кг детям, далее — 0,5 и 0,25 г/кг соответственно. Болюсное введение препарата уменьшает гемоконцентрацию и продлевает действие маннитола [30]. Максимальный эффект препарата достигается примерно через 60 мин, а общая продолжительность действия может достигать 4—6 ч. При непрерывном введении эффективность маннитола снижается, так как происходит увеличение вязкости крови. Это приводит к снижению ЦПД и, согласно механизму ауторегуляции мозгового кровотока, к вазодилатации церебральных сосудов и повышению ВЧД. Вторичное повышение ВЧД происходит также из-за так называемого эффекта «отдачи», когда постоянное повышение концентрации препарата в крови ведет к его перемещению по осмотическому градиенту в ткань (внеклеточное пространство), где он накапливается, создавая «обратный» осмотический градиент. Жидкость из сосудистого русла по «обратному» осмотическому градиенту устремляется во внеклеточное пространство, увеличивая его объем — развивается отек мозга, ведущий к повышению ВЧД.
При неадекватном и длительном применении маннитола возможно развитие осложнений. К ним относят: гиперосмолярное (преренальное) поражение почек вплоть до развития почечной недостаточности; электролитные нарушения (гипернатриемия и гипокалиемия); дегидратация и артериальная гипотензия, повышающие риск церебральной ишемии (снижение ЦПД), который выше у пожилых пострадавших и у пациентов с сочетанными повреждениями. Маннитол может потенцировать развитие интрацеребрального кровоизлияния.
В связи с этим препаратом выбора для проведения осмодиуретической терапии для борьбы с ВЧГ при тяжелой ЧМТ, особенно у больных с шоком, сочетанной травмой в последнее время является гипертонический раствор NaCl в 3—3,5% концентрации у детей и 7% — у взрослых в дозировке — 0,1 г/кг массы тела. Ряд авторов рекомендуют его сочетание с альбумином или растворами гидроксиэтила крахмала [36, 42].
Другим лекарственным средством борьбы с ВЧГ являются барбитураты (тиопентал натрия). Несмотря на то, что этот препарат прошел серию доказательных исследований, показавших его эффективность для борьбы с ВЧГ, его применение ограничено строгими рамками показаний. Эффективность барбитуратов обусловлена многими механизмами нейропротективного действия, к которым относят: снижение церебрального метаболизма; перераспределение регионарного мозгового кровотока; снижение ВЧД; супрессию судорожной гиперактивности; блокаду терморегуляции; стабилизацию мембран нейронов; блокаду определенных мембранных каналов; угнетение метаболизма жирных кислот; барбитураты являются «ловушкой» свободнорадикальных соединений. Тем не менее их применение сопровождается развитием многочисленных осложнений, способных нивелировать положительные эффекты препарата и даже ухудшить состояния пациента. К ним относят: артериальную гипотензию, которая встречается у 58% больных в условиях барбитуровой защиты; анергию, обусловливающую скрытое развитие инфекции; развитие трофических нарушений и тромбоза вен; развитие пареза желудочно-кишечного тракта; высокий риск кардиологических осложнений; быстрое формирование водно-электролитных нарушений. К осложняющему фактору относят и невозможность клинической оценки состояния больного в течение 12 и более ч после прекращения введения барбитуратов.
Поэтому барбитураты в настоящее время используют как часть протокола поэтапного снижения ВЧД, когда неэффективны другие методы интенсивной терапии, а у детей — не используются совсем [34, 37, 38]. Показания к их применению являются обоснованием к декомпрессивной трепанации черепа, как крайней мере церебропротективного воздействия.
C позиций доказательной медицины эффективность при травматических субарахноидальных кровоизлияниях показал нимодипин. Его действие достоверно улучшало исход у пациентов до 40 лет мужского пола. Являясь антагонистом Са2+-каналов (L- типа), хорошо проникая через гематоэнцефалический барьер из-за выраженной липофильности он доказал свою эффективность при церебральном вазоспазме при САК травматического и нетравматического генеза. Помимо этого в ряде исследований было показано его прямое противоишемическое и антиоксидантное действие. Препарат применяется в виде внутривенной инфузии в суточной дозе от 30 мг на протяжении 5—10 суток. Это, с одной стороны, определяет его быстрый эффект, с другой, может приводить к развитию фактора вторичного повреждения мозга — острой артериальной гипотензии (см. выше). Ее предупреждение и коррекция поддержанием объема циркулирующей крови и/или дозированным введением вазопрессоров гарантирует конечные позитивные эффекты препарата [20, 29, 39, 41].
Не прошли этап доказательных исследований, но, тем не менее, применяются в остром периоде церебральных повреждений разнообразные лекарственные средства избирательного действия (антагонисты глютаматных рецепторов, антиоксиданты, макроэрги и т.д.). Назначая их, следует помнить о возможном антагонизме действия препаратов на различных уровнях: системном, органном, рецепторном и др. Необходимо учитывать вероятность конкуренции за одни и те же рецепторы, наконец, имеет смысл иметь в виду правило «пяти препаратов», которое всегда является актуальным для пациента в крайне тяжелом, критическом состоянии. В связи со всем этим, выбор «дополнительного» лекарственного препарата должен быть обоснован, в том числе его «особым» вариантом фармакологического действия.
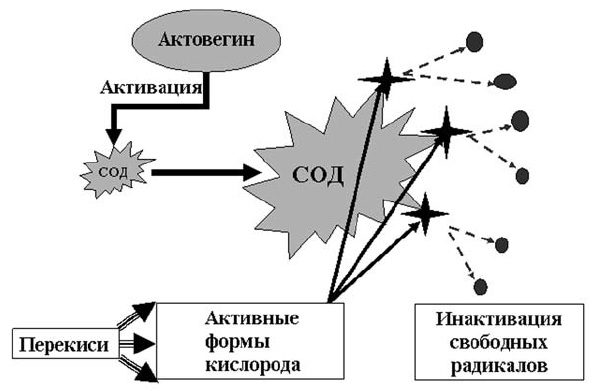
Если говорить об антиоксидантах, то традиционно мы представляем этот класс препаратов, как лекарственные средства замещающего действия, эффект которых обусловлен связыванием свободнорадикальных соединений. Механизм субстратного взаимодействия тесно связан и зависит от количества препарата, его достаточной субстратной представленности, а какова она на самом деле, в клинических условиях, как правило, мы не представляем. В этой связи целесообразно напомнить о том, что существует так называемый «физиологический» путь фармакологического воздействия на процессы перекисного окисления. Таков механизм действия актовегина, который запускает естественный антиоксидантный механизм в организме человека через активацию супероксиддисмутазы, которая, в свою очередь являясь ключевым ферментом процессов перекисного окисления, связывает и инактивирует избыток свободнорадикальных соединений (схема 10).
Схема 10. Физиологический путь антиоксидантного действия актовегина.
CОД — супероксиддисмутаза.Такой механизм действия предполагает необходимость введения в организм пациента не насыщающее (субстратное), а предрасполагающее («запускающее» какой-либо процесс) количество препарата, что соответствует терапевтической дозировке актовегина.
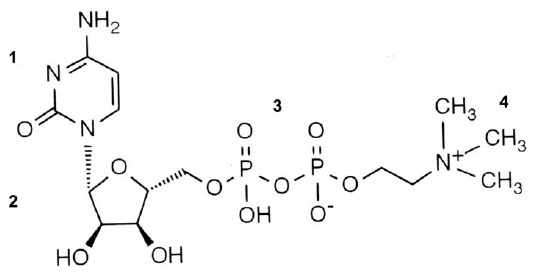
В связи с упоминанием лекарственных средств, прошедших доказательные исследования своей эффективности, следует отметить долгожданное появление в нашем арсенале такового под названием цераксон. Действующее его вещество — цитиколин (цитидин-5-дифосфохолин, CDP-холин, ЦДФ-холин) является незаменимым предшественником фосфатидилхолина (лецитина), основного структурного компонента всех клеточных мембран, включая нейрональные мембраны. А его структурная формула (рис. 11) во многом объясняет многочисленные нижеперечисленные фармакологические эффекты, в ней присутствуют и нуклеотидная составляющая, и СН3-группы, и двойные мостики связей, и многое другое.
Рис. 11. Структурная формула цераксона.
1 — нуклеотид, 2 — рибоза, 3 — дифосфатный мостик, 4 — холин.К эффектам цераксона относят: активацию биосинтеза фосфатидилхолина (цитидин-5дифосфохолин); поддержание нормоуровня кардиолипина и сфингомиелина; прямое участие в синтезе ацетилхолина; стимуляцию синтеза глутатиона; ингибирование процессов перекисного окисления липидов (антиоксидантные свойства); восстановление активности Na+ — K+ — АТФ-азы; угнетение активности фосфолипазы А2; снижение глутаматиндуцированного апоптоза; активацию энергетических процессов в нейронах; активацию цитохромоксидаз в митохондриях нейронов (нормализация процессов тканевого дыхания).
Цераксон — один из немногих препаратов, выдержавших требования доказательной медицины, и с успехом применяющийся более 10 лет в странах Европы и в США [21—23].
Следует учесть, что цераксон был целенаправленно создан и специально предназначен для защиты и восстановления нервной ткани при ее ишемии (нарушения мозгового кровообращения при инсультах, ЧМТ, дисциркуляторная энцефалопатия различного генеза). Он входит в стандарты лечения ишемического инсульта в ряде стран Европы. Имеет дозозависимый эффект в виде уменьшения зоны пенумбры; комплексно воздействует на ЦНС: оказывает поддерживающее и восстанавливающее (регенераторно-репаративное) структурное, оптимизирующее метаболические и энергетические процессы, нейромедиаторный обмен, улучшающее синаптическую передачу, тормозящее каскадные процессы и образование свободнорадикальных соединений действие. Цераксон предназначен для применения на всех этапах лечения, включая острейший период травмы и инсульта, где клинические эффекты его применения наиболее наглядны. А с учетом доказанной безопасности препарата для пациентов это, в свою очередь, предполагает правомочность планирования клинических исследований эффективности цераксона при его применении в первые часы после церебральных повреждений и даже превентивно, при наличии риска развития церебральной ишемии и гипоксии, то есть на догоспитальном этапе, еще до уточнения диагноза церебрального повреждения инструментальными методами, но при наличии его клинических проявлений. С учетом имеющейся информации о препарате есть уверенность в том, что он может и должен войти в перечень средств, рекомендуемых с целью нейропротекции уже при первых симптомах развития церебрального поражения. Наличие пероральной и парентеральной форм делают удобным использование препарата у всех категорий амбулаторных и стационарных больных. Биологическая доступность препарата практически равна 100%, будучи аналогом эндогенного цитиколина, препарат на 84% включается в обменные процессы, прежде всего в нервной ткани [18, 26]. Особого обсуждения, как с позиций научного исследования, так и клинического воздействия, требует ожидаемый синергизм лечебных эффектов при курсовом лечении препаратами цераксон и актовегин у всех категорий амбулаторных и стационарных больных. При их подробном рассмотрении мы можем наблюдать редко встречаемый случай, когда взаимодополняющие эффекты препаратов предполагают сильное позитивное воздействие на организм больного в критическом состоянии, при патологических состояниях различного генеза. Описан синергизм лечебного воздействия при «упреждающем» применении цераксона в комбинации реперфузионной терапии с рекомбинантным тканевым активатором плазминогена (rTPA) при остром ишемическом инсульте. Но это предмет отдельного рассмотрения с позиций будущих клинических исследований.
Хирургический подход
Этот подход к церебропротекции предусматривает комплекс лечебных мероприятий, предупреждающих или уменьшающих действие внутричерепных факторов вторичного повреждения мозга в рамках хирургического вмешательства. Речь идет об установке вентрикулярного дренажа при синдроме нарастающей ВНГ, имплантации датчика для измерения внутричерепного давления, осуществлении декомпрессивной трепанации черепа. Нейропротективными являются хирургические вмешательства, направленные на эвакуацию из полости черепа дополнительно образовавшихся объемов внутричерепного содержимого (эпидуральные, субдуральные, внутримозговые скопления крови, значительные контузионные очаги), вызывающих развитие синдромов сдавления мозга и дислокации мозговых структур. Нейропротективным вмешательством, с точки зрения хирургического пособия, является и проведение реперфузии и реканализации при развитии острой ишемии мозговой ткани (ОНМК), а также выполнение анастомозирующих (шунтирующих) операций. В настоящее время большинство этих нейропротективных вмешательств входят в международные рекомендации по лечению пациентов с тяжелой ЧМТ и церебральным инсультом в качестве соответствующих строгих протоколов и требуют отдельного рассмотрения.
1 В случаях управляемой гипотермии, управляемой артериальной гипотензии и гипертензии, а также гиперволемии, гемоделюции, гипервентиляции и гипергликемии есть основания поставить знак вопроса.
2 КО рассчитывается по формуле КО=V2/V1, где V1 — средняя величина линейной скорости мозгового кровотока (ЛСК МК), полученная при проведении фонового исследования методом транскраниальной допплерографии (ТКД);. V2 — средняя величина ЛСК МК первого — второго пиков после прекращения компрессии общей сонной артерии (пальцевым надавливанием в области шеи).
Литература
- Амчеславский В.Г., Брагина Н.Н., Мадорский С.В. и др. Особенности формирования критических состояний у нейрохирургических больных в послеоперационном периоде. В сб.: Интенсивная терапия острых нарушений мозгового кровообращения. Орел 1997; 182—185.
- Брагина Н.Н., Смирнова Н.Я., Хухлаева Е.А. Основные принципы клинического исследования послеоперационных состояний в нейрохирургии. Сборник научных работ: Интенсивная терапия, реанимация и анестезия в нейрохирургии. М 1982.
- Бунятян А.А., Рябов Г.А., Маневич А.З. Анестезиология и реаниматология. Учебное пособие. М: Медицина 1977; 430.
- Данелия Т.З. Оценка и обеспечение энергетических потребностей у нейрохирургических больных, требующих интенсивную терапию: Автореф. дис. ... канд. мед. наук. М 1986.
- Зильбер А.П. Клиническая физиология для анестезиолога. М: Медицина 1977; 431.
- Кассиль В.Л., Руда М.Я. Руководство по интенсивной терапии (в клинике внутренних болезней). М: Медицина 1976; 224.
- Корниенко В.Н., Потапов А.А., Пронин И.Н., Захарова Н.Е. Диагностические возможности компьютерной и магнитно-резонансной томографии при черепно-мозговой травме. Гл. 13, с.408-464., В кн: Доказательная нейротравматология. Под ред. А.А. Потапова. М 2003.
- Маневич А.З., Салалыкин В.И. Нейроанестезиология. М: Медицина 1977; 320.
- Маневич А.З., Потапов А.А., Брагина Н.Н. Патоморфологические основы интенсивной терапии тяжелой черепно-мозговой травмы. Вестн АМН СССР 1986; 6: 60—64.
- Мухаметжанов X. Внутричерепная гипертензия в остром периоде тяжелой ЧМТ: Автореф. дис. ... канд.мед. наук. М 1987; 25.
- Неговский В.А., Гуревич А.М., Золотокрылов Е.С. Постреанимационная болезнь. М: Медицина 1987; 480.
- Потапов А.А. Патогенез и дифференцированное лечение очаговых и диффузных повреждений головного мозга: Автореф. дис. … д-ра мед. наук. М 1989; 54.
- Потапов А.А., Лихтерман Л.Б., Зельман В.Л. и др. Доказательная нейротравматология. М 2003.
- Пясецкая М.В. Автореф. дис. ... канд. мед. наук. М 1978; 28.
- Рябов Г.А. Критические состояния в хирургии. М 1979; 318.
- Сафин А.М. Автореф. дис. ... канд. мед. наук. М 1987; 25.
- Сировский Э.Б., Амчеславский В.Г., Данелия Т.З. и др. Обоснование принципов интенсивной терапии в нейрохирургии. Вестн интенсивной тер 1992; 1.
- Adibhatla R.M., Hatcher J.F., Dempsey R.J. Citicoline: neuroprotective mechanisms in cerebral ischemia. J Neurochem 2002; 80: 1: 12—23.
- Albin M.S. Textbook of Neuroanesthesia with Neurosurgical and Neuroscience Perspectives. The McGraw-Hill Companies Inc., 1997.
- Bailey I., Bell A., Gray J. et al. A trial of the effect of nimodipine on outcome after head injury. Acta Neurochir (Wien) 1991; 110: 3—4: 97—105.
- Clark W.M., Warach S.J., Pettigrew L.C. et al. A randomized dose-response trial of citicoline in acute ischemic stroke patients. Citicoline Stroke Study Group. Neurology 1997; 49: 671—678.
- Clark W.M., Williams B.J., Selzer K.A. et al. A randomized efficacy trial of citicoline in patients with acute ischemic stroke. Stroke 1999; 30: 2592—2597.
- Clark W.M., Wechsler L.R., Sabounjian L.A., Schwiderski U.E. Citicoline Stroke Study Group. A phase III randomized efficacy trial of 2000 mg citicoline in acute ischemic stroke patients. Neurology 2001; 57: 1595—1602.
- Cruz J. An additional therapeutic effect of adequate hyperventilation in severe acute brain trauma: normalization cerebral glucose uptake. J Neurosurg 1995; 82: 379—385.
- DeSalles A.F., Muizelaar J.P., Young H.F. Hyperglycemia, cerebrospinal fluid lactic acidosis and cerebral blood flow in severely head-injured patients. Neurosurgery 1987; 21: 45—50.
- D’Orlando K.J., Sandage B.W.Jr. Citicolone (CDP-choline): mechanisms of action and effects In ischemic brain injuri. Neurol Res 1995; 17: 281—284.
- Gisvold S.-E. The Lund concept for treatment of head injuries — faith or science? Acta Anaesthesiol Scand 2001; 45: 399—401.
- Guidelines for the Management of severe head injury, Brain Trauma Foundation, 1995.
- Harders A., Kakarieka A., Braakman R. and German tSAH Study Group: Traumatic subarachnoid hemorrhage and its treatment with nimodipine. J Neurosurg 1996; 85: 82—89.
- James H.E. Methodology for the control of intracranial pressure with hypertonic mannitol. Acta Neurochirurgica 1980; 51: 161—172.
- Management and Prognosis of Severe Traumatic Brain Injury 2000. Brain Trauma Foundation 2000.
- Marion D.W., Penrod L.E., Kesley S.F. et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med 1997; 336: 540—546.
- Muizelaar J.P., Marmarou A., Ward J.D. et al. Adverse effects of prolonged hyperventilation in patient with severe head injury: A randomized clinical trial. J Neurosurg 1991; 75: 731—739.
- Nordstrom G.H., Messeter K., Sundberg B. et al. Cerebral blood flow, vasoreactivity and oxygen consumption during barbiturate therapy in severe traumatic brain lesions. J Neurosurg 1988; 68: 424—431.
- Obrist W.D., Langfitt T.W., Jaggi J.L. et al. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg 1984; 61: 241—253.
- Pascual J.M.S., Runyon D.E., Watson J.C. et al. Resuscitation of hypovolemia using near saturated sodium chloride solution in Dextran. Circ Shock 1993; 40: 115—124.
- Rea G.L., Rockswold G.L. Barbiturate therapy in uncontrolled intracranial hypertension. Neurosurgery 1983; 12: 401—405.
- Rockoff M.A., Marshall L.F., Shapiro H.M. High-dose barbiturate therapy in humans: a clinical review of 60 patients. Ann Neurol 1979; 6: 194—199.
- Roine R.O. et al. Nimodipine after resustitacion from out-of-hospital ventricular fibrillation: A placebo-controlled, double-blind randomized trial. JAMA 1990; 264: 3171.
- Rosner M.J., Rosner S.D., Johnston A.H. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg 1995; 83: 949—962.
- The European Study Group on Nimodipine in Severe Head Injury: A multicenter trial of the efficacy of nimodipine on outcome after severe head injury. J Neurosurg 1994; 80: 797—804.
- Vassar M.J., Fischer R.P. et al. A multicenter trial for resuscitation of injured patients with 7.5 % sodium chloride/ the effect of added dextran 70. The Multicenter Group for the Study of Hypertonic Saline in Trauma Patients. Arch Surg 1993; 128: 1003—1011.
- Warach S., Pettigrew L.C., Dashe J.F. et al. Effect of citicoline on ischemic lesions as measured by diffusion-weighted magnetic resonance imaging. Citicoline 010 Investigators. Ann Neurol 2000; 48: 713—722.
| Апрель 2010 г. |